
Las enfermedades raras son enfermedades potencialmente mortales o, crónicamente debilitantes, que se caracterizan por ser poco comunes. Como resultado, los medicamentos huérfanos (MMHH), dirigidos a tratarlas, se enfrentan a una serie de dificultades específicas para su comercialización y seguimiento.
El objetivo de este artículo es analizar en qué consisten los problemas a los que se enfrentan los medicamentos huérfanos para lograr su autorización comercial, así como las distintas soluciones que se han planteado en la práctica para superarlos. Asimismo, se exponen las pautas de comercialización de los medicamentos huérfanos, y se tratan los registros de pacientes como una de las iniciativas más utilizadas en la fase de seguimiento post-autorización de este tipo de fármacos.
LA EVIDENCIA CLÍNICA EN EL ÁMBITO DE LOS MEDICAMENTOS HUÉRFANOS
La autorización de todo medicamento corre a cargo de la Agencia Europea del Medicamento (EMA), que exige la demostración de su eficacia, seguridad y calidad, en base a estudios clínicos. Estos criterios se aplican también a los MMHH, si bien, dadas sus características y problemáticas asociadas, se les permite cierta laxitud sobre la evidencia aportada (se les exige datos sobre su seguridad y plausibilidad, en vez de sobre su seguridad y eficacia).
Las enfermedades raras son enfermedades potencialmente mortales o crónicamente debilitantes.
Normalmente, en un experimento clínico estándar, el número de sujetos analizados debe ser lo suficientemente grande como para alcanzar una diferencia en los resultados clínicos que sea estadísticamente significativa1. Para calcular el tamaño muestral se debe tener en cuenta la frecuencia esperada del desenlace en el grupo de interés y en el grupo control, la probabilidad de que ocurra un error aleatorio y la diversidad de la población a estudiar, requiriéndose menos sujetos si la enfermedad tiene un comportamiento más homogéneo.
Sin embargo, realizar un ensayo clínico para un medicamento huérfano supone dificultades de diversa índole. Dada la baja prevalencia de la enfermedad a tratar, la muestra estará previsiblemente compuesta por pocos sujetos. Por lo tanto, habrá una alta probabilidad de que la muestra no sea representativa de la población a estudiar, especialmente si existe una elevada variabilidad entre los sujetos. Además, la significación estadística de los resultados puede verse afectada, no pudiendo demostrarse un efecto que realmente existe. Aplicar una metodología de calidad es importante para que los resultados del estudio tengan validez interna.
A esto se une la dispersión geográfica de los pacientes, la falta de un comparador apropiado y el escaso conocimiento sobre la enfermedad (su epidemiología, patofisiología, historia natural y farmacología, elementos necesarios para diseñar estudios clínicos eficientes)2. Por estas razones, los estudios clínicos en enfermedades raras no siempre logran el nivel de evidencia que alcanzan los estudios con muestras más grandes y pacientes más uniformes.
Además, las dificultades de los ensayos clínicos con MMHH se extienden a dilemas éticos y al reclutamiento de pacientes3. En enfermedades graves que carecen de tratamiento, administrar placebo a los sujetos implica importantes problemas éticos, ya que se pone en riesgo su vida a pesar de existir un producto que muy probablemente mejoraría su esperanza de vida. Por ello, debe tratar de minimizarse el tamaño del grupo de control y, asegurarse de que, una vez concluido el estudio, también se les administrará el fármaco a testar si se demostró su efectividad4. Por su parte, el reclutamiento de pacientes aislados, geográficamente dispersos y no registrados, puede resultar difícil.
Así, a menudo se acabará tratando a ambas ramas del estudio clínico con el medicamento huérfano en cuestión, lo que elevará el coste promedio del mismo, que a su vez será de por sí superior al de un fármaco convencional, por tener que reclutar a pacientes que se encuentran repartidos en distintos centros, lejanos geográficamente entre sí, y que exigen esfuerzos proactivos de reclutamiento. Otro inconveniente tiene que ver con la dificultad de extraer conclusiones sobre la efectividad relativa del medicamento a medio y largo plazo9.
Por estos motivos, en la fase de pre-autorización, la generación de evidencia clínica de un medicamento huérfano puede ser aceptable a través de estudios menos exigentes que los ensayos clínicos aleatorizados, como por ejemplo ensayos no aleatorizados o estudios observacionales donde la comparación se hace con datos históricos.

EL NIVEL DE EVIDENCIA DEL PARACAÍDAS
El caso del paracaídas, para el cual sólo se han realizado estudios observacionales y no ensayos clínicos aleatorizados frente a placebo, sirve como un ejemplo extremo para ilustrar que en situaciones de peligro inminente de muerte, las intervenciones no necesitan aportar un nivel de evidencia alto para justificar su uso5. En el ámbito sanitario, este símil puede aplicarse a intervenciones como la apendicectomía o la cesárea, en casos de sufrimiento fetal agudo6. Podría ser trasladado también a los casos de enfermedades raras más graves para las que no existen alternativas terapéuticas eficaces, como por ejemplo el síndrome de Hutchinson-Gilford, entre otras muchas7,8.
Se han propuesto distintas soluciones para tratar de paliar el problema del reducido tamaño muestral que sufren los MMHH, que pasan por la elección de un diseño alternativo o por el uso de variables de resultado alternativas (por ejemplo, subrogadas). Algunos autores también proponen ajustar los ensayos estándares, por ejemplo eligiendo una mayor duración del estudio para captar un mayor número de eventos10 o centrarse en los sujetos con más riesgo11.
Elección de variables de resultado
Una forma de reducir los requerimientos de tamaño muestral de los estudios con MMHH es a través de la selección de variables de resultado o endpoints alternativas, tales como una variable que sea continua en vez de binaria, una variable compuesta que combine múltiples variables, o una variable subrogada o intermedia.
Una variable subrogada se mide en lugar de la variable clínicamente relevante, generalmente porque es más fácil de medir o de identificar, pudiendo ambas tener una relación directa, o sólo indirecta. La opción de utilizar variables subrogadas como solución intermedia para generar evidencia clínica es una opción que la EMA reconoce como aceptable, siempre que su uso esté justificado. Debe existir una relación clara con la eficacia clínica, de modo que sea posible evaluar los riesgos y beneficios12.
La opción de utilizar variables subrogadas como solución intermedia para generar evidencia clínica es una opción que la EMA reconoce como aceptable.
Estudios de diseños alternativos
La EMA reconoce que, cuando no sea posible obtener evidencia controlada sobre la eficacia y seguridad de un nuevo tratamiento, es posible aceptar enfoques alternativos al gold standard, siempre que éstos aseguren la protección de los intereses de los pacientes, elaborando una guía sobre diseños alternativos para n pequeñas10. La guía concluye que la necesidad de disponer de eficiencia estadística debe ponderarse por la necesidad de disponer de resultados clínicamente relevantes e interpretables, siendo esto último lo más importante. Estos y otros diseños alternativos propuestos en la literatura se resumen en la Tabla 1, que recoge además algunos ejemplos reales aplicados a MMHH. El objetivo principal de estos enfoques es o bien minimizar el número total de participantes o bien minimizar la muestra tratada con placebo y el tiempo de tratamiento inactivo3,13.
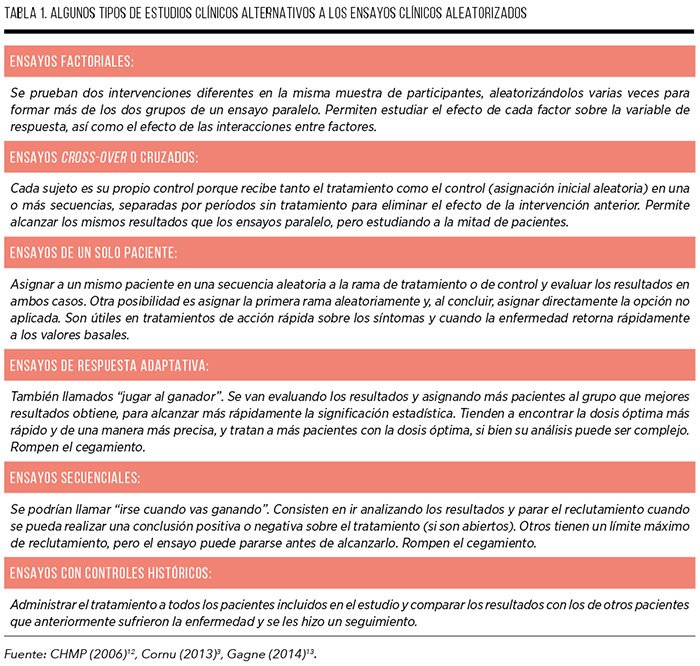
Estas alternativas permiten sortear algunas de las principales limitaciones de los ensayos clínicos estándar aplicados a medicamentos huérfanos, como son no alcanzar la significación estadística, no encontrar suficientes
pacientes que cumplan con los criterios de inclusión o el dilema ético de no tratar a pacientes con una dolencia grave. Otras ventajas son que permiten ahorrar tiempo (ensayo factorial), optimizar el tamaño muestral (ensayo cruzado), encontrar más rápidamente la dosis precisa (ensayo adaptativo), minimizar el tiempo en tratamiento inefectivo (ensayo adaptativo) o potenciar la adherencia (ensayo de un solo paciente).
No obstante, también presentan algunos inconvenientes de los que hay que ser conscientes. Los diseños secuenciales y adaptativos incumplen el criterio de cegamiento, incrementan la complejidad del análisis y aumentan la probabilidad de obtener conclusiones espurias14. En los ensayos cruzados y de un solo paciente la enfermedad debe ser estable, y el estado de salud inicial debe ser similar al comenzar ambas fases de tratamiento, lo cual puede no producirse en enfermedades raras. Asimismo, pese al período de blanqueo*, los efectos del primer período pueden persistir al comenzar el segundo período de tratamiento aleatorio, dificultando la extrapolación de conclusiones. Finalmente, muchos diseños sirven para evaluar respuestas en un período corto de tiempo, dejando sin explorar los efectos a medio y largo plazo.
* Se entiende por periodo de blanqueo o lavado aquel que se produce entre dos intervenciones consecutivas aplicadas a un mismo paciente, con el fin de que desaparezcan los posibles efectos de la primera intervención y los efectos de la segunda puedan ser exclusivamente adjudicados a esta intervención y no a una mezcla de la dos.
El argumento central de las excepciones de evidencia que se permiten a los medicamentos huérfanos es que, desde un punto de vista social, es defendible que los pacientes que sufren una enfermedad rara tengan derecho a percibir el mismo acceso a un tratamiento seguro y efectivo que los pacientes que sufren enfermedades comunes o prevalentes15.
Habitualmente, la calidad de los estudios de evidencia para muestras pequeñas se evalúa a través de marcadores individuales, listas de comprobación y escalas. Recientemente se ha desarrollado y validado un nuevo instrumento, el COMPASS (Clinical evidence of Orphan Medicinal Products – an ASSessment tool), para evaluar la calidad de la evidencia presentada por los MMHH en el momento de la autorización de comercialización en la UE16.
El COMPASS se utilizó para evaluar la calidad de la evidencia clínica presentada por 64 MMHH (en 117 estudios) al solicitar la autorización de comercialización17. El análisis puso de manifiesto que en el 69% de los estudios se utilizó una rama de control; la asignación de pacientes fue aleatoria en el 65%; el 50% aplicó algún tipo de cegamiento; el 49% comprobó que la población de estudio era representativa de la población general; y sólo el 27% incluyó una variable de resultado relacionada con la calidad de vida.
AUTORIZACIÓN COMERCIAL DE MEDICAMENTOS HUÉRFANOS
La designación de un medicamento como huérfano no garantiza que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización por parte de la EMA. Estos criterios sólo pueden ser evaluados una vez que se ha presentado la solicitud de autorización de comercialización.
En el caso de los MMHH, el proceso de autorización comercial sólo puede obtenerse mediante procedimiento centralizado y se realiza de manera individualizada, con decisiones caso a caso, para tratar de conciliar el acceso rápido al tratamiento huérfano con la garantía de que dicho tratamiento sea efectivo y seguro.
La autorización de comercialización de un medicamento por parte de la EMA puede ser de 3 tipos, en función del nivel de evidencia aportado18,19:
• Autorización normal o estándar, cuando se aporta evidencia comprehensiva sobre la eficacia, seguridad y calidad del medicamento en cuestión. Su validez es de 5 años.
• Autorización condicional. En determinadas categorías de medicamentos, para satisfacer necesidades no cubiertas de los pacientes y en interés de la salud pública, puede concederse una autorización basada en datos menos completos de lo habitual (evidencia temprana) que indiquen un previsible balance beneficio/riesgo positivo del fármaco. La autorización es solo temporal, y debe renovarse anualmente hasta que se hayan completado los estudios necesarios para evaluar la autorización normal.
• Autorización bajo circunstancias excepcionales. Si el solicitante puede demostrar una serie de razones objetivas y verificables (baja prevalencia, limitado conocimiento científico o consideraciones éticas) por las que no puede aportar evidencia sobre la eficacia y seguridad del fármaco, puede otorgarse una autorización temporal bajo circunstancias excepcionales, revaluable anualmente. El solicitante debe asegurar la seguridad del producto y notificar cualquier incidente al respecto.
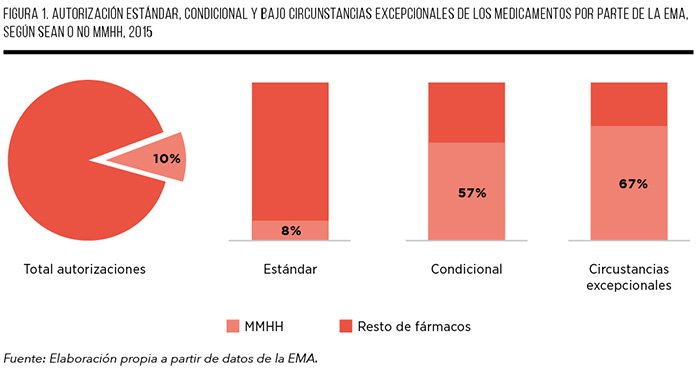
Los medicamentos huérfanos constituyen el núcleo de las autorizaciones excepcionales concedidas por la EMA. Así, aunque suponen sólo el 10% del total de medicamentos actualmente autorizados, representan el 57% de las autorizaciones condicionales y el 67% de las autorizaciones bajo circunstancias especiales actualmente vigentes, con 8 y 16 fármacos, respectivamente, en estas situaciones de autorización (Figura 1)20.
Una vez autorizado el fármaco a nivel europeo, cada país decide si autorizarlo o no en su territorio, y a qué precio. Así, no todos los medicamentos huérfanos tienen por qué registrarse en todos los países. Por ejemplo, de los 87 MMHH autorizados a nivel europeo, sólo 51 (un 59%) están comercializados en España (Figura 2)21,22.

Para decidir la comercialización del medicamento, las autoridades sanitarias de varios países europeos, entre ellos España, se valen de los Informes de Posicionamiento Terapéutico (IPTs). En España, los IPT son coordinados por la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), y pretenden establecer un sistema de evaluación en red basado en la evidencia científica que evite redundancias y mantenga la coherencia en la evaluación entre los distintos agentes implicados, compartiendo los recursos de la forma más eficiente posible23.
Los informes ofrecen información relevante, basada en la evidencia científica, sobre la posición que el nuevo medicamento ocupa en el mercado en comparación con otros medicamentos o medidas de salud ya existentes, estableciendo su grado de aportación terapéutica. Han de servir como una de las bases para la financiación selectiva y, en su caso, fijación de precios de los medicamentos de uso humano. Hasta 2015, aproximadamente una cuarta parte de los IPTs realizados en España se referían a MMHH24.
Por último, cabe señalar que las Comunidades Autónomas y los hospitales son quienes finalmente deciden qué medicamentos se administran en los hospitales. Para ello, se valen de distintos tipos de guías terapéuticas para facilitar la selección del tratamiento. Los medicamentos huérfanos también pueden ser sometidos a criterios de posicionamiento terapéutico, para ajustar la indicación y el seguimiento de los pacientes de forma precisa25. También se les puede requerir evidencia adicional sobre su eficacia, seguridad o conveniencia, incluso aunque hayan sido aprobados por la EMA bajo la autorización estándar.
REGISTROS DE PACIENTES
Como hemos visto, en el momento de la comercialización de un medicamento huérfano a menudo existe muy poca información para analizar el balance riesgo-beneficio del fármaco, lo que hace indispensable la existencia de un programa de seguimiento post-autorización. Una de las mejores formas de abordarlo es a través de la creación de registros clínicos de pacientes, que permitan realizar un seguimiento prospectivo del fármaco en pacientes del “mundo real”, en contraste con el “mundo ideal” de los estudios experimentales26.
Los registros de pacientes aportan información muy valiosa sobre la epidemiología y el curso natural de la enfermedad. Pero, además, son esenciales para monitorizar la seguridad y efectividad de los tratamientos a medio y largo plazo, reclutar más pacientes en estudios experimentales y observacionales, encontrar variables predictivas de diagnóstico y sustentar estudios de farmacovigilancia12,27. Asimismo, permiten poner en contacto a los pacientes y reducir así su sensación de aislamiento.
En general, existen tres tipos de registros:
- Para pacientes que comparten una misma enfermedad o condición.
- Para pacientes que comparten la exposición a una misma intervención o grupo de medicamentos.
- Para pacientes que comparten ciertas características generales (por ejemplo, unos rasgos físicos similares). Para ser de mayor utilidad, los registros deben contener información estandarizada, completa y actualizada de los casos tanto tratados, como no tratados27. Idealmente estarán gestionados por un organismo independiente14. Los registros de pacientes con EERR se han popularizado en los últimos años. Actualmente en Europa hay unos 520 registros de enfermedades raras, 490 de ellos manejados por entidades universitarias6. En España existe, desde 2005, un Registro Nacional de Enfermedades Raras, que pretende recoger información de todas las personas diagnosticadas de cualquier enfermedad considerada rara o de baja prevalencia. Con esta herramienta se pretende mantener un censo de pacientes, conocer la incidencia y prevalencia de las enfermedades raras, facilitar la comparación entre regiones y orientar la planificación y gestión sanitaria, con el objetivo de contribuir al desarrollo de nuevos tratamientos y a la mejora de la prevención, diagnóstico, pronóstico y calidad de vida de los pacientes. En diciembre de 2015 se aprobó el Real Decreto que lo regula28, y actualmente se está trabajando en su desarrollo, a través de la redacción de su manual de procedimiento.
No sólo se han creado registros individuales en el ámbito de las enfermedades raras, sino también distintas redes, consorcios, plataformas y repositorios, a nivel tanto nacional como europeo, para aglutinar registros y fomentar la colaboración, generando así mayores sinergias. En la Tabla 2 se recogen los logotipos de algunas de estas iniciativas.
Actualmente en Europa hay unos 520 registros de enfermedades raras, 490 de ellos manejados por entidades universitarias.
Finalmente, cabe destacar el proyecto EUROPLAN (Proyecto Europeo para el Desarrollo de Planes Nacionales para las Enfermedades Raras), cuyo propósito principal es impulsar la creación armonizada de planes y estrategias de enfermedades raras en la UE29. En la fase dos del proyecto EUROPLAN (2012-2015) se han celebrado 24 Conferencias Nacionales para tratar los desafíos experimentados durante la adopción e implementación de los planes nacionales.

En el contexto del EUROPLAN, en España se conformaron 10 Grupos de Trabajo, uno de los cuales estaba focalizado en consolidar el Registro Nacional y la Red Española de Registros para la Investigación de Enfermedades Raras, así como en mejorar la interoperabilidad entre los registros de pacientes ya existentes. El Registro Nacional presenta la doble vertiente de registro poblacional y registro de pacientes específicos para determinadas enfermedades raras. Su desarrollo se ha fortalecido a través de acuerdos con todas las CCAA, para incorporar todos los casos detectados, de la incorporación de registros específicos de enfermedades concretas y del proyecto “Red Española de Registros para la Investigación de EERR (SpainRDR)”, que establece una metodología común para garantizar la armonización de la recogida de datos a nivel autonómico y nacional30.
CONCLUSIONES
Dada la baja prevalencia de las enfermedades raras, los medicamentos huérfanos se enfrentan a dificultades de diversa índole cuando tratan de evidenciar su eficacia y seguridad. Realizar ensayos clínicos con MMHH a menudo implica una baja representatividad de la muestra, así como dilemas éticos y dificultades de reclutamiento. Por ello, en la fase de pre-autorización se les aplican ciertas exenciones, como son la aportación de evidencia basada en estudios menos exigentes que los ensayos clínicos aleatorizados, o el uso de variables subrogadas.
Asimismo, las autoridades europeas a menudo permiten autorizar la comercialización de un fármaco huérfano, de manera temporal, basada en evidencia temprana, en aras de acelerar el acceso de los pacientes al tratamiento. Posteriormente, son los estados miembro quienes deciden la comercialización del medicamento en cada país, para lo que se valen, entre otros, de los IPTs.
Una de las mejores formas de abordar el seguimiento post-autorización de un medicamento huérfano es a través de los registros de pacientes. Los registros aportan información muy valiosa sobre la seguridad y efectividad del fármaco, pero también sobre la epidemiología y el curso natural de la enfermedad, resultando esenciales para mejorar el conocimiento de la enfermedad, el reclutamiento de pacientes y el contacto entre las familias. Durante los últimos años, se han dado distintos pasos, tanto en Europa como en España, para optimizar y mejorar la interoperabilidad de los registros y redes de pacientes con enfermedades raras.
Dada la baja prevalencia de las enfermedades raras, los medicamentos huérfanos se enfrentan a dificultades de diversa índole cuando tratan de evidenciar su eficacia y seguridad
REFERENCIAS
- Centers for Medicare & Medicaid Services. Outcome-Based Quality Improvement (OBQI) Manual. (2010).
- Paz, MP et al. in Rare Diseases Epidemiology 17–39 (Springer Netherlands, 2010).
- Cornu, C. et al. Experimental designs for small randomised clinical trials: an algorithm for choice. Orphanet J Rare Dis 8, 48 (2013).
- Van der Lee, J. H., Wesseling, J. & Tanck, M. W. T. Efficient ways exist to obtain the optimal sample size in clinical trials in rare diseases. Journal of clinical epidemiology 61, 324–330 (2008).
- Smith, G. C. S. & Pell, J. P. Parachute use to prevent death and major trauma related to gravitational challenge: systematic review of randomised controlled trials. BMJ 327, 1459–1461 (2003).
- Rosselli, D & Rueda, JD. Enfermedades raras, huérfanas y olvidadas. (2011).
- Orphanet. Papulosis atrófica maligna. Available at: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=679.
- Orphanet. Síndrome de Hutchinson Gilford. Available at: http://www.orpha.net
- Muñoz, SR & Bangdiwala, SI. Análisis interino en ensayos clínicos: una guía metodológica. Revista médica de Chile 128, 935–941 (2000).
- Shurin, S., Krischer, J. & Groft, S. C. Clinical Trials In BMT: Ensuring That Rare Diseases and Rarer Therapies are Well Done. Biology of Blood and Marrow Transplantation 18, S8–S11 (2012).
- Whitehead, J., Tishkovskaya, S. & O’Connor, J. Devising two-stage and multistage phase II studies on systemic adjuvant therapy for uveal melanoma. Invest Ophthalmol Vis Sci 53, 4986–4989 (2011).
- Committee for Medicinal Products for Human Use (CHMP) & De Lorenzo. Guideline on Clinical Trials in Small Populations. (Springer, 2006).
- Gagne, J. J., Thompson, L., O’Keefe, K. & Kesselheim, A. S. Innovative research methods for studying treatments for rare diseases: methodological review. The BMJ 349, g6802 (2014).
- Garjon, J. Medicamentos huérfanos: regulación y controversias. Boletín de Información Farmacoterapéutica de Navarra 33, (2015).
- Schieppati, A., Henter, J.-I., Daina, E. & Aperia, A. Why rare diseases are an important medical and social issue. The Lancet 371, 2039–2041 (2008).
- Picavet, E, Cassiman, D, Aertgeerts, B & Simoens, S. Development and validation of COMPASS: clinical evidence of orphan medicinal products – an assessment tool. Orphanet J Rare Dis 8, (2013).
- Picavet, E., Cassiman, D., Hollak, C. E., Maertens, J. A. & Simoens, S. Clinical evidence for orphan medicinal products-a cause for concern? Orphanet J Rare Dis 8, 164 (2013).
- Regulation No 726/2004 of the European Parliament and of the Council, laying down the Community procedures for the authorization and supervision of medicinal products. Official Journal EU 136,
- Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use (2001).
- European Medicines Agency – Find medicine – European public assessment reports.
- AEMPS – Centro de Información online de Medicamentos de la AEMPS (CIMA).
- AEMPS. Propuesta de colaboración para la elaboración de los informes de posicionamiento terapéutico de los medicamentos. Doc. aprobado por la Comisión Permanente de Farmacia del SNS.
- Agencia Española de Medicamentos y Productos Sanitarios – Medicamentos de uso humano – Informes de posicionamiento terapéutico. Available at: http://www.aemps.gob.es.
- Puigventós, F, Calderón, B & Gorgas, MQ. Posicionamiento de los medicamentos en guías terapéuticas y protocolos clínicos. (SEFH. Ediciones Mayo, 2009).
- Luisetti, M. et al. The problems of clinical trials and registries in rare diseases. Respiratory medicine 104, S42–S44 (2010).
- Campillo, C & Peiró, S. Enfermedades raras, medicamentos huérfanos: el valor de la orfandad. GCS 42, 119–127 (2010).
- Real Decreto 1091/2015, de 4 de diciembre, por el que se crea y regula el Registro Estatal de Enfermedades Raras.
- Eurordis Rare Disease Europe. Reports of EUROPLAN National Conferences 2012-2015.
- EUCERD Joint Action. EuroPlan España. Informe Final de la Conferencia Nacional. (2014).







