María Teresa Marín Rubio
Directora General de Humanización y Atención Sociosanitaria de la Consejería de Sanidad de Castilla-La Mancha
Castilla-La Mancha ha apostado decididamente por la humanización en la atención sanitaria. ¿Podría contarnos cuáles son las principales líneas estratégicas del Plan de Humanización en la comunidad y cómo se integran en la atención a pacientes con enfermedades raras?
MTM: El Plan de Humanización de la asistencia sanitaria de Castilla-La Mancha se aprobó en el año 2022, con horizonte 2025. Nos encontramos en un momento de plena implantación.
Yo diría, en primer lugar, que uno de los aspectos más importantes de este plan es la metodología que hemos utilizado para elaborarlo y que seguimos utilizando en su implantación. Se trata de una metodología muy participativa, basada en la inteligencia colectiva, con espacios de cocreación y grupos de trabajo multidisciplinares muy activos e implicación de la sociedad. De esta manera, desde su propio origen, el plan es algo muy vivo y participativo.
Los vectores estratégicos de este plan serían, en primer lugar, la participación. En segundo lugar, el enfoque de todas las medidas hacia el cuidado de los y las profesionales, así como una especial atención a los espacios físicos y confort.
Con este plan queremos superar el trabajo que veníamos desarrollando desde antes de 2015 en materia de humanización, centrado sobre todo en los detalles del cuidado: el “plan de los pequeños detalles”, el “Plan Dignifica”. Queremos ir más allá de esas medidas locales y puntuales, y orientar nuestro enfoque hacia un cambio profundo en la cultura de la organización. El plan entiende la humanización como un proceso, un proceso en el que se despliegan una serie de valores que deben respaldar, una atención sanitaria más centrada en la persona, en sus familiares y cuidadores, y también en los profesionales.
La idea de que la humanización es un proceso, y no simplemente un conjunto de medidas que modifican aspectos puntuales de la asistencia nos lleva a incluir como estratégico ese cambio cultural dentro de la organización. La estructura y cultura de Humanización es otro de sus vectores de desarrollo.
Y, por último, y cerrando todos estos vectores de desarrollo del plan de humanización, está la atención integral centrada en la persona. Es cierto que este concepto puede parecer ya algo desgastado, pero sigue siendo útil, especialmente cuando hablamos, como vamos a hacer hoy, del reto de la atención a personas con una enfermedad rara o poco frecuente. En estos casos, quizás la dificultad más importante o, mejor dicho, el principal desafío, sea precisamente representar el paradigma de lo que debe ser la atención centrada en la persona. Si pensamos en una persona con una enfermedad rara, veremos que concentra todos esos elementos que luego pueden desarrollarse también en otras patologías, pero que en el caso de las enfermedades raras o poco frecuentes aparecen de manera más grave, más intensa.
Los determinantes sociales son un eje clave en la salud y el bienestar. ¿Qué papel juegan en los planes y políticas de humanización de Castilla-La Mancha?
MTM: Si hablamos del Plan de Humanización como un cambio cultural, como una verdadera cultura de la humanización, en esa visión estratégica y transversal que debe impregnar a toda la organización, los determinantes sociales tienen un papel fundamental.
Desde el año 2015, en Castilla-La Mancha venimos insistiendo en la necesidad de un cambio de modelo. Un cambio que desplace el foco desde la atención sanitaria entendida exclusivamente como respuesta a los problemas de salud, hacia un concepto más amplio: la atención a la salud, incluso antes de que esos problemas aparezcan. Si sustituimos el concepto de “sanidad” por el de “salud”, no queda más remedio que hablar de determinantes sociales.
Hablamos, por tanto, de desigualdades, de desarrollo sostenible, de los entornos que nos rodean, de todo aquello que influye, y que es causa y consecuencia, de lo que después se manifiesta como un problema de salud o como un enfoque poco adecuado del bienestar de las personas.
Los determinantes sociales adquieren una relevancia especial en aquellos grupos de población más vulnerables. Podemos hablar de personas con bajos ingresos, pero también de personas con trastornos mentales, de pacientes crónicos, o de colectivos afectados por la despoblación. En una comunidad como Castilla-La Mancha, el envejecimiento poblacional ya no es el único reto; se suma la enorme dispersión geográfica, que implica que muchas personas de edad muy avanzada vivan en localidades o en entornos muy aislados, lo que añade una gran dificultad tanto para atender sus problemas de salud como para abordar su contexto vital.
Esos determinantes sociales hacen que muchas personas accedan a los recursos sanitarios en condiciones de desigualdad. Y, una vez más, esto tiene una especial importancia en colectivos como el de las personas que padecen una enfermedad rara o poco frecuente.
Por lo tanto, en nuestro enfoque de humanización y en nuestro modelo de atención a la salud de la población de Castilla-La Mancha, los determinantes sociales son fundamentales. El sistema sanitario y la atención que prestamos se ven afectados por estos determinantes, pero también pueden convertirse, si no actuamos con la debida sensibilidad, en origen de nuevas desigualdades o barreras para determinados colectivos.
Es decir, si la asistencia que ofrecemos no presta especial atención a la equidad, a la justicia social y a los derechos humanos, corremos el riesgo de generar más inequidad con nuestras propias intervenciones.
La vida humana es holística, resultado de muchas circunstancias distintas, y evoluciona a lo largo del ciclo vital. Nosotros debemos acompañar a las personas durante todo ese proceso. Si no tenemos en cuenta el medio ambiente, natural, animal, económico y social, y cómo nuestras intervenciones en salud lo afectan, difícilmente podremos construir ese enfoque innovador, integral, transversal y longitudinal que queremos aplicar para cuidar la salud de la ciudadanía.
Además, es fundamental que esa atención se preste en el entorno en el que vive el ciudadano. Esto implica reforzar la atención primaria, impulsar la estrategia de salud comunitaria, que ya está en marcha en Castilla-La Mancha y tener presente nuestra estrategia de lucha contra la despoblación, en la que fuimos pioneros y que ha demostrado ser muy exitosa. Esta estrategia también debe formar parte del enfoque integral de salud.
Una de las grandes barreras que enfrentan las personas con enfermedades poco frecuentes es la falta de equidad en el acceso a servicios y recursos. ¿Cómo se trabaja desde su Dirección General para reducir estas desigualdades y garantizar una atención más justa, inclusiva y centrada en la persona?
MTM: Pasando de lo más general a lo más concreto, podemos aterrizar en un caso particular. Cuando hablamos de equidad, nos referimos a un trato justo, a dar a cada cual lo que le corresponde. Esto implica reconocer y conocer las condiciones, características y necesidades específicas de cada persona. Solo así podemos evitar situaciones de discriminación.
¿Cómo trabajar, entonces, desde ese enfoque? Como decíamos antes, las enfermedades raras representan quizás el paradigma de la necesidad de una atención más humanizada, porque estamos hablando de equidad y justicia, pero teniendo en cuenta las necesidades concretas de este colectivo.
En primer lugar, hablamos de participación y colaboración con la iniciativa social. En Castilla-La Mancha, el Plan de Humanización se desarrolla en estrecha colaboración con el tejido asociativo vinculado a las enfermedades raras. Este movimiento asociativo tiene unas características particulares: debido a la baja prevalencia de estas enfermedades, las asociaciones están formadas por un número reducido de personas, lo que les resta peso y visibilidad.
Por ello, estas asociaciones necesitan trabajar desde un modelo de colaboración, formando alianzas que les permitan ser interlocutores válidos ante la administración y defender de manera más amplia las necesidades y demandas del colectivo. En Castilla-La Mancha llevamos años trabajando junto a estas asociaciones, generando alianzas, elaborando guías, estableciendo criterios para convocatorias de ayudas y subvenciones, ofreciendo formación y desarrollando iniciativas como la Escuela de Salud y Cuidados.
Todo esto lo hacemos de manera directa desde nuestra Oficina de Atención a Enfermedades Raras, que realiza tanto el acompañamiento y asesoramiento puntual a las familias como una labor de seguimiento, colaboración y coordinación con la iniciativa social. Esta oficina ofrece un apoyo específico, que resulta imprescindible en un ámbito como este, donde la participación social es más reducida debido al bajo número de personas afectadas.
En cuanto a los y las profesionales, sabemos que necesitan formación e información actualizada y fácilmente accesible. Es fundamental que puedan establecer contacto ágil con especialistas. En muchos casos, algunos facultativos, tanto en atención primaria como hospitalaria, nunca han atendido a un paciente con una enfermedad rara concreta, debido a su bajísima incidencia. Puede haber un único paciente en toda Castilla-La Mancha con una determinada patología.
Nuestros profesionales deben tener acceso a la información y a herramientas que les permitan contactar con unidades de experiencia o con los CSUR (Centros, Servicios y Unidades de Referencia) que sí disponen del conocimiento específico. En este sentido, Castilla-La Mancha participa en el programa UNICAS, con dos hospitales ya integrados. Este programa facilita la interconsulta entre centros y permite que la información circule entre profesionales, evitando que siempre tenga que desplazarse el paciente, salvo que sea estrictamente necesario.
Uno de los principales problemas que denuncian los pacientes es que tienen que explicar una y otra vez su situación y enfrentarse al desconocimiento de los profesionales que los atienden en cada consulta. Evitar esto es uno de nuestros compromisos y está alineado con nuestra línea de trabajo centrada en el cuidado y la formación de los y las profesionales.
También trabajamos en la mejora de los espacios. Ya hemos habilitado en muchos de nuestros hospitales espacios específicos para consultas multidisciplinares dirigidas a personas con enfermedades raras. Este año, además, hemos impartido un curso intensivo de formación para enfermeras gestoras de casos, una figura clave para acompañar a la persona y su familia en su tránsito por el sistema sanitario.
Cuidamos tanto el espacio físico como el espacio organizativo: el diseño de protocolos de derivación, la ordenación de citas, la coordinación entre profesionales… Todo ello forma parte de un espacio de atención que debe estar bien estructurado para facilitar la experiencia del paciente. La enfermera gestora de casos es esencial en este modelo, y por eso hemos intensificado su formación y acreditación.
Por último, en relación con la atención integral y centrada en la persona, desde la Oficina de Enfermedades Raras acompañamos de forma directa a las personas afectadas y sus familias. Estamos presentes en momentos clave como el diagnóstico, el agravamiento de la enfermedad, las crisis o los cuidados paliativos. Lo hacemos en colaboración con el movimiento asociativo, cuando existe, y también desde un enfoque profesional, ofreciendo apoyo psicológico desde la propia Oficina, que depende de la Dirección General de Humanización.
Trabajamos para que este acompañamiento sea cercano, oportuno y respetuoso, contribuyendo a una atención verdaderamente humanizada que tenga en cuenta las particularidades de cada persona, cada patología y cada contexto familiar.
El bienestar emocional y el apoyo psicosocial son esenciales para las personas con enfermedades raras y sus familias. ¿Qué recursos contempla el sistema castellano-manchego para reforzar estos aspectos dentro de una atención humanizada?
MTM: Nosotros creemos que, además de contar con psicólogos y profesionales específicamente formados, lo fundamental es impulsar un cambio cultural que implique a toda la organización. Estamos trabajando en la sensibilización en torno a estas patologías, no solo en el ámbito sanitario, sino también en otros entornos clave.
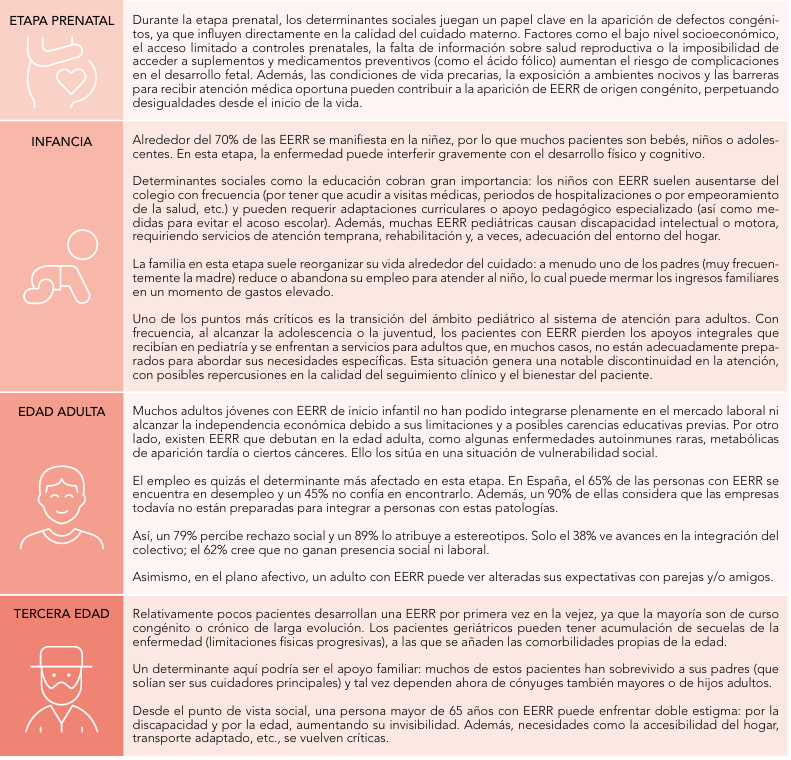
Un ejemplo claro es el ámbito educativo. Muchas de estas enfermedades se diagnostican en la infancia, por lo que es fundamental que el sistema educativo esté preparado para atender, integrar y cuidar a estos niños y niñas en su entorno. Por ello, hemos llevado a cabo varios cursos de formación dirigidos a profesionales de la educación, con el objetivo de fomentar una mayor integración y comprensión.
Una de nuestras líneas estratégicas fundamentales es la formación y sensibilización de todos los profesionales, no solo los sanitarios. También desarrollamos numerosas campañas dirigidas a la ciudadanía en general, para fomentar una mayor conciencia social sobre las enfermedades raras.
Además, estamos trabajando en colaboración con el Instituto de Investigación de Castilla-La Mancha (IRICYS) para reforzar las líneas de investigación y promover la inversión en este campo. La investigación es siempre el futuro, y esto es especialmente relevante en el caso de las enfermedades raras.
La colaboración con las asociaciones de pacientes también resulta clave. No solo mejora la calidad de la atención, sino que permite un acompañamiento más cercano en momentos críticos. En muchos casos, es la propia persona afectada, o un familiar que ha vivido la enfermedad de cerca, quien está en mejor disposición de prestar apoyo emocional y psicológico a otras familias que atraviesan situaciones similares.
Este apoyo psicológico se presta en distintos niveles: desde el movimiento asociativo, en nuestras consultas hospitalarias y de atención primaria, y también de forma transversal en cualquier ámbito asistencial, a través de una formación continua en humanización de la asistencia.
Cuando hablamos de colectivos con necesidades especiales, como las personas con enfermedades poco frecuentes, enfermedades crónicas o trastornos mentales graves, es imprescindible reforzar la sensibilización y el acompañamiento. No se trata solo de atención médica, sino de ofrecer un apoyo integral a la persona y a su familia, entendiendo el impacto emocional y social que conlleva convivir con estas patologías.
En el marco del modelo de atención centrado en la persona, ¿cómo se está promoviendo la participación activa de pacientes, cuidadores y asociaciones de pacientes en el diseño de servicios y en la toma de decisiones sanitarias?
MTM: Desde el inicio, estamos trabajando en un modelo muy participativo, que parte del propio diseño de los programas. Es decir, en todos los planes y estrategias que hemos puesto en marcha recientemente, o que lo harán en breve, la participación es un eje fundamental.
Hablamos de la participación de la ciudadanía, de los pacientes, de los profesionales, de los directivos… En todos los niveles (macro, meso y micro), la implicación comienza ya en la fase de diseño. Esto no solo facilita la involucración, sino que permite construir políticas más ajustadas a las necesidades reales.
Pero la participación no se agota en el hecho de invitar a colaborar: requiere también de un feedback real y sistemático, una devolución transparente de los resultados obtenidos en cada proceso. Para ello, estamos utilizando herramientas tecnológicas y metodológicas que nos permiten mantener ese canal abierto de retroalimentación durante todo el proceso de planificación.
Este enfoque lo estamos aplicando, por ejemplo, en la Estrategia de Salud Comunitaria, en la que trabajamos desde hace tiempo, así como en otras actualmente en desarrollo, como la Estrategia de Daño Cerebral o la Estrategia de Enfermedades Raras, que verá la luz muy pronto.
Además, la participación es una de las líneas estratégicas más destacadas de nuestro Plan de Humanización, hasta el punto de haber generado un laboratorio de innovación específico sobre participación. Precisamente ayer presentamos públicamente este plan en Valdepeñas, bajo el nombre de Salud y Sociedad. Se trata de un programa absolutamente centrado en la participación social de la ciudadanía, entendiendo que la salud, como servicio público esencial, debe gestionarse también con una mirada democrática. Este plan busca que la ciudadanía participe de forma activa en la toma de decisiones, en la definición de prioridades y en la orientación de la inversión en salud. Salud y Sociedad ya está en marcha y está llamado a ser uno de los planes estrella, porque recoge numerosas medidas concretas. Entre ellas, destaca la necesidad de dimensionar qué tipo de entidades deben participar, cómo y en qué momentos. No se trata solo de abrir espacios de participación, sino de definir bien esos espacios según los niveles en los que se opera.
Por ejemplo, a nivel micro, hablamos de la consulta, de la escucha activa, del papel del paciente en la toma de decisiones clínicas, o de su formación como paciente experto, algo que impulsamos desde la Escuela de Salud y Cuidados.
A nivel meso, trabajamos con las Gerencias de Atención Integrada, que son la estructura más común en Castilla-La Mancha, para facilitar y estructurar espacios de participación efectivos.
Y a nivel macro, estamos promoviendo medidas que transformarán el papel de la ciudadanía en el diseño de las políticas sanitarias.
Estoy convencida de que todas estas acciones supondrán un revulsivo en la intensidad, el impacto y la calidad de la participación, y que su repercusión será muy significativa.
Si decimos que el enfoque de nuestro Plan de Humanización es poner a la persona en el centro, entonces la participación de esa persona debe ser el instrumento clave para lograrlo. El programa Salud y Sociedad alimentará y a la vez se nutrirá de este plan, modificando por completo las herramientas de participación que veníamos utilizando hasta ahora. Es un paso más hacia la innovación y la adaptación a las nuevas formas de participación social.
Hoy en día, con las redes sociales y las nuevas tecnologías, ya no hay excusa: la participación puede darse casi en tiempo real. Y el feedback también. Nuestro reto, y nuestra responsabilidad, es gestionar bien esa información, procesarla y devolverla convertida en acciones concretas, en servicios y prestaciones adecuados a lo que la ciudadanía realmente necesita.
Por eso, Salud y Sociedad no es solo un programa, es una apuesta firme por una participación real, significativa y transformadora, que sitúa a la ciudadanía en el corazón de las decisiones sobre su propia salud.
Sabemos que la coordinación sociosanitaria es fundamental en enfermedades de curso crónico o complejo. ¿Qué avances destacaría en la articulación entre el sistema sanitario y los servicios sociales para mejorar la calidad de vida de estos pacientes?
MTM: Actualmente estamos trabajando en el desarrollo del nuevo Plan de Atención Sociosanitaria de Castilla-La Mancha, que formará parte del marco del Plan de Salud vigente, cuyo horizonte es 2025. Aunque ese horizonte marca el final formal del plan actual, ya estamos definiendo su revisión y la siguiente etapa, porque evidentemente la planificación no se detiene en 2025. Dentro del plan Horizonte 2025 ya se incluía el compromiso de contar con un plan específico de atención sociosanitaria en nuestra comunidad.
No está siendo fácil. Esta es una dificultad compartida con otras comunidades autónomas y también en los foros de coordinación ministerial. En el ámbito sociosanitario intervienen múltiples actores: educación, servicios sociales, vivienda, salud… Y la coordinación entre todas estas áreas requiere una estructura sólida y estable.
En ese sentido, el nuevo plan de coordinación sociosanitaria incluirá estructuras permanentes de coordinación, que consideramos imprescindibles. Uno de los grandes retos es definir un catálogo claro de prestaciones sociosanitarias, porque no contar con uno dificulta enormemente cualquier avance. Saber qué prestaciones se ofrecen, a quién, en qué condiciones y cómo se financian es un paso clave. Y ese catálogo aún no está cerrado. Por eso, los retos del nuevo plan no son solo conceptuales ni metodológicos, sino también financieros y estructurales.
Entre los desafíos que tenemos por delante destaca, por supuesto, la atención al colectivo de personas con enfermedades raras, pero también la salud mental, que es un reto cada vez más urgente. No solo en lo relativo al trastorno mental grave, sino también al malestar emocional que estamos detectando con más frecuencia y a edades cada vez más tempranas, lo cual es especialmente preocupante.
También estamos poniendo el foco en colectivos como las personas con discapacidad o quienes viven con enfermedades crónicas, muchas de ellas asociadas al envejecimiento de la población. Este es un fenómeno que podemos anticipar y que va a tener un gran impacto en los próximos años.
Nuestra intención es que el nuevo Plan de Atención Sociosanitaria dé respuesta a todos estos desafíos. Pero para garantizar la equidad, también esperamos una mayor coordinación desde el Ministerio, que ayude a asegurar el acceso equitativo a los recursos sociosanitarios en todo el territorio nacional, y a desarrollar líneas comunes de actuación que favorezcan la igualdad real entre comunidades autónomas.
En Castilla-La Mancha ya hemos dado pasos importantes en esta dirección. Hace tiempo pusimos en marcha un acuerdo marco de colaboración entre la Consejería de Bienestar Social, la Consejería de Educación y la Consejería de Sanidad. Este acuerdo nos ha permitido avanzar de manera muy significativa en temas relacionados con el entorno educativo. Por ejemplo, hemos trabajado intensamente en la prevención del acoso escolar, el ciberacoso, y en salud sexual y reproductiva. También se ha hecho un esfuerzo importante en la prevención del suicidio, donde contamos con un marco estratégico que ha dado muy buenos resultados en la comunidad.
Además, se ha avanzado en formación y se está trabajando mucho en el desarrollo de protocolos de continuidad de cuidados, un elemento esencial para una atención sociosanitaria efectiva y centrada en la persona.
Finalmente ¿qué mensaje le gustaría transmitir sobre el valor de humanizar la atención a las personas con enfermedades raras y sobre los próximos retos que debemos afrontar en este ámbito?
MTM: Si hablamos de futuro, yo creo que la esperanza está en la capacidad que tienen las sociedades para enfrentarse a los retos que se les presentan. Por supuesto, hay que trabajar mucho, y lo estamos haciendo, pero confío en esa capacidad colectiva que hemos demostrado a lo largo de la historia. Y, cuando hablamos de futuro, la esperanza siempre está ligada a la innovación.
Es cierto que en esta entrevista hemos hablado poco de innovación, y muchas veces, cuando lo hacemos, tendemos a asociarla únicamente con la investigación biomédica, con nuevos medicamentos, nuevas tecnologías clínicas, nuevas herramientas terapéuticas. Y es verdad, esa innovación es imprescindible. Pero no es la única.
La innovación más transformadora, la que más impacto tiene sobre la vida de las personas, es la innovación social. Y ahí es donde estamos centrando una parte muy importante de nuestro trabajo: en humanizar la existencia desde una óptica de innovación social. Esto significa que todo lo que hacemos en el sistema sanitario, toda la atención que prestamos a la ciudadanía va a estar orientado a mejorar su bienestar y su salud desde una mirada innovadora y profundamente humana.
Queremos que la atención a la salud en Castilla-La Mancha tenga ese enfoque de humanización como eje vertebrador, que todo pase por ese filtro de valores. Hablamos de una atención sanitaria: corresponsable, que implique a todos los actores; sostenible, para que perdure en el tiempo y comprometida con los derechos humanos, porque no puede haber salud sin equidad ni dignidad. Y, sobre todo, queremos una atención que nos prepare para afrontar cualquier reto futuro, desde una mirada centrada en las personas.
La humanización desde la innovación social. Ese sería mi mensaje.






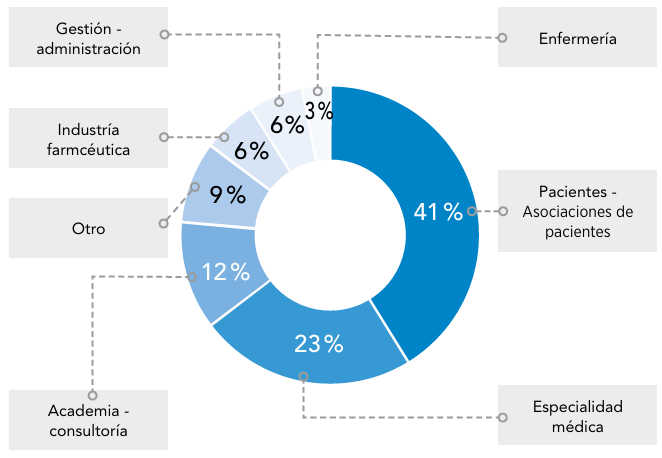
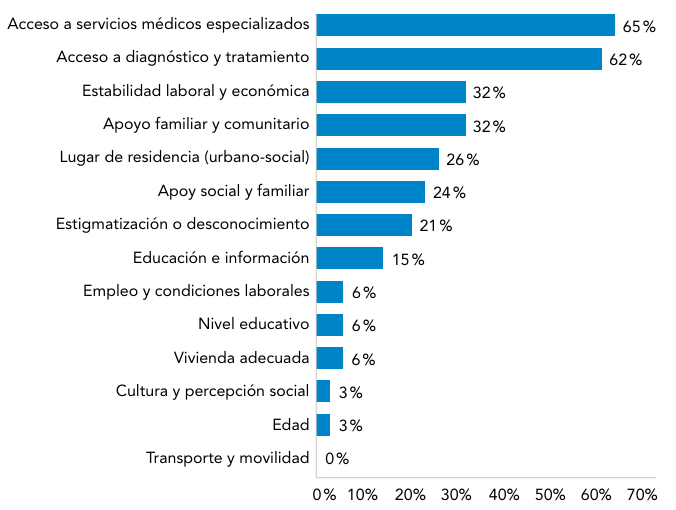
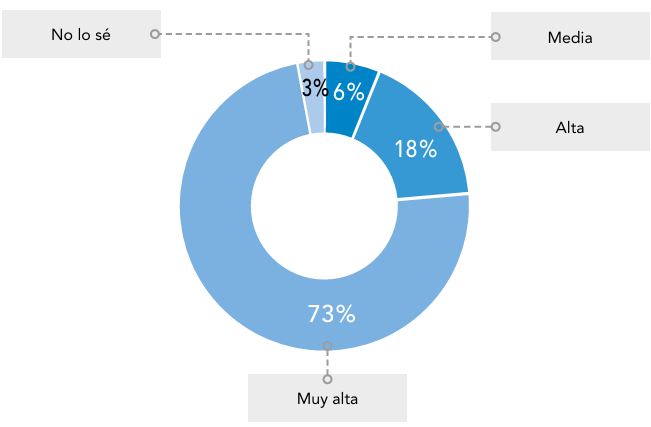
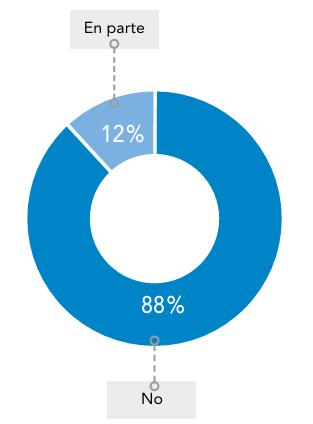
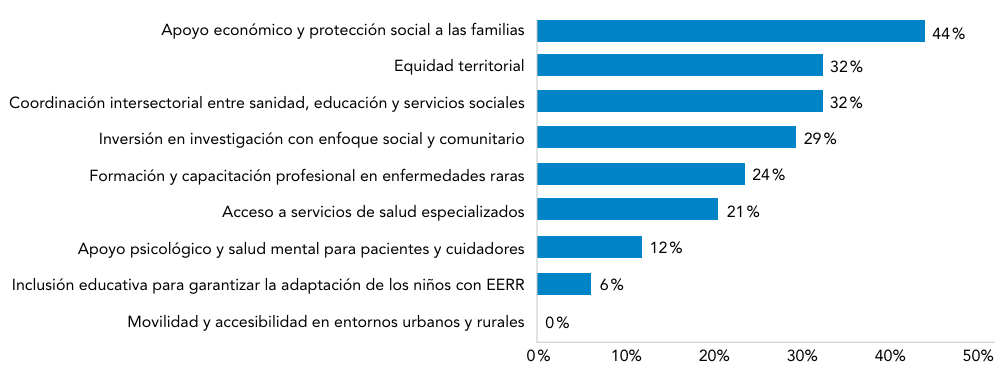
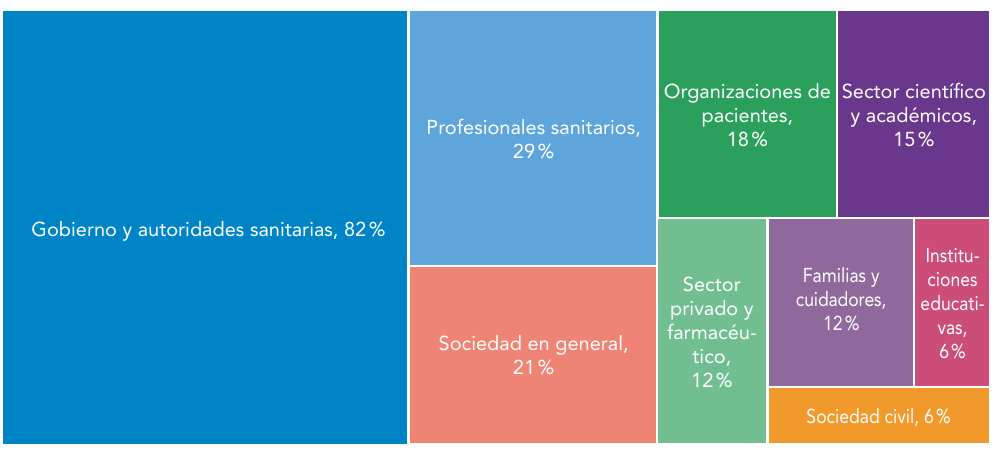
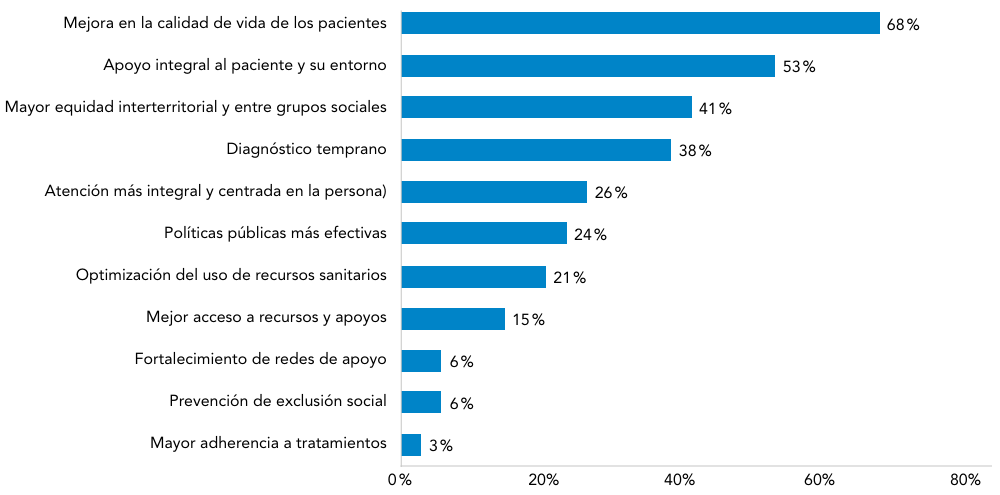
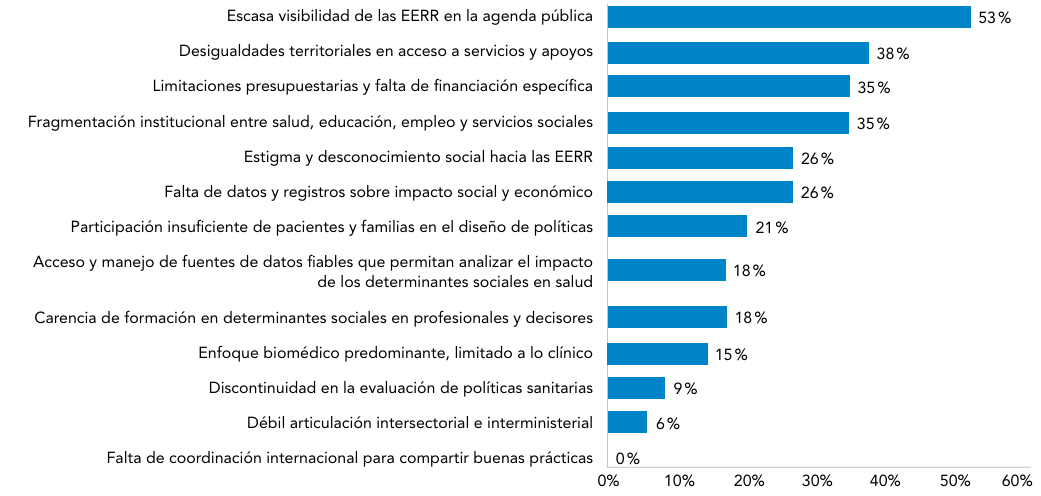
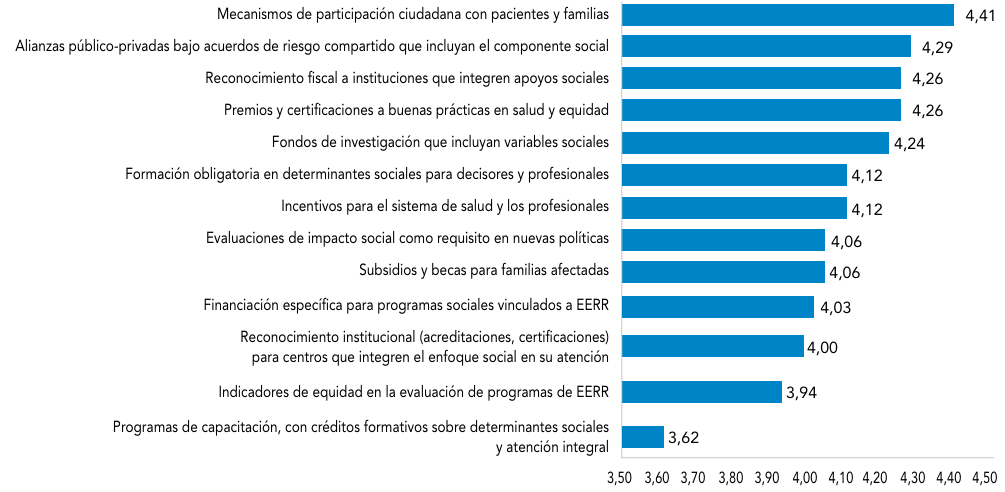

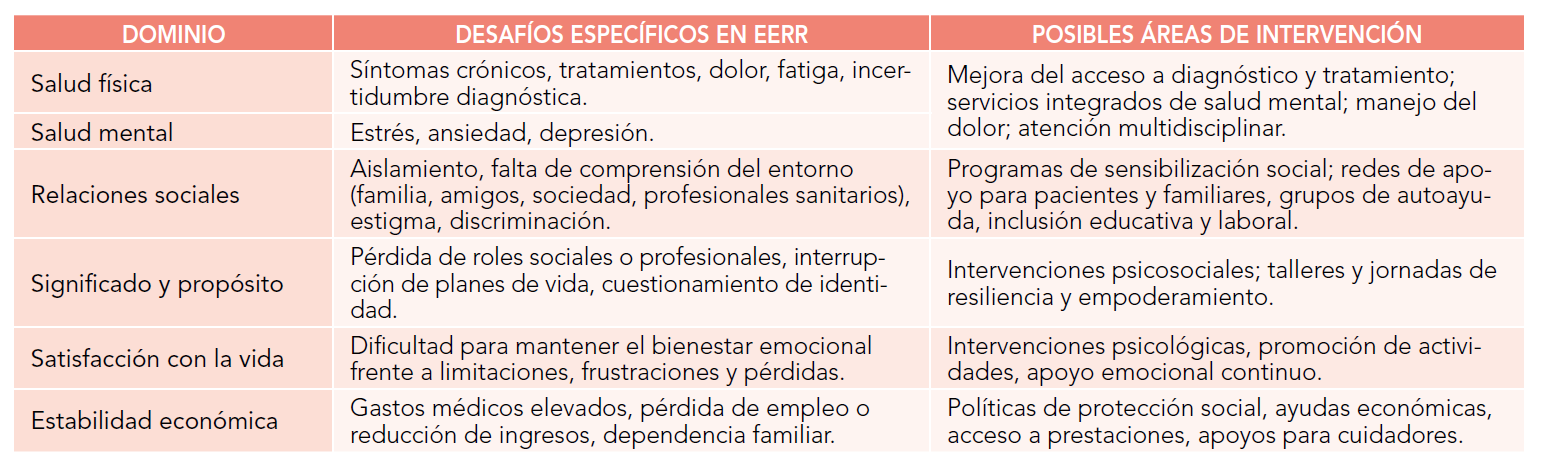
 Abreviaturas: EERR: enfermedades raras.
Abreviaturas: EERR: enfermedades raras.  Abreviaturas: EERR: enfermedades raras.
Abreviaturas: EERR: enfermedades raras. 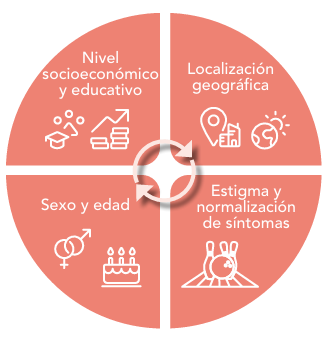 Abreviaturas: EERR: enfermedades raras.
Abreviaturas: EERR: enfermedades raras. 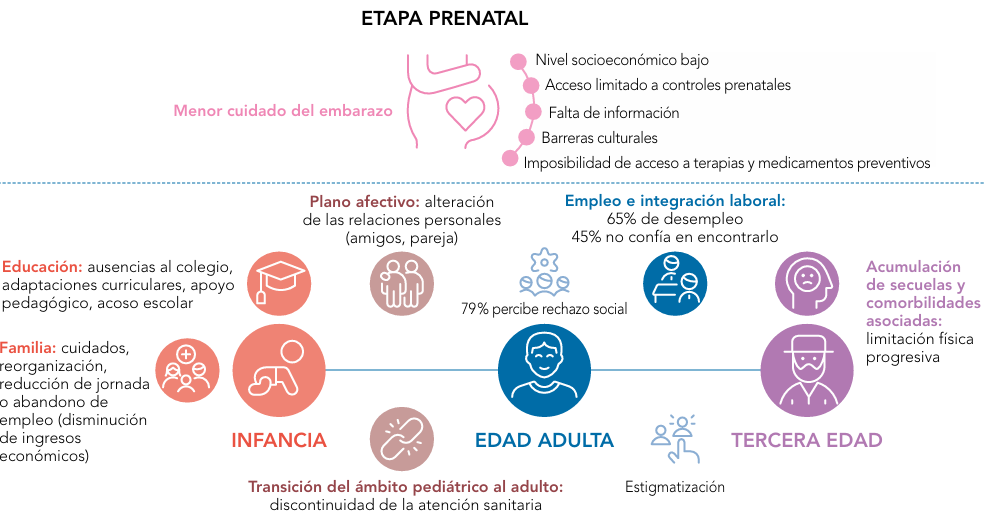
 INICIATIVAS Y PERSPECTIVAS DE MEJORA
INICIATIVAS Y PERSPECTIVAS DE MEJORA