Néboa Zozaya y Bleric Alcalá
El proceso de precio y reembolso de los medicamentos huérfanos (MMHH) es complejo y controvertido. Por una parte, las restricciones presupuestarias exigen un uso eficiente de los recursos limitados y el establecimiento de prioridades. Por otra parte, establecer el valor real que aportan los MMHH no es fácil, dada la incertidumbre asociada a la evolución de la patología, la escasez de evidencia y de alternativas terapéuticas con las cuales comparar su beneficio clínico. Asimismo, la baja frecuencia de la enfermedad, que en muchos casos se caracteriza por ser grave, progresiva, crónica e incapacitante, con sus correspondientes consecuencias clínicas, económicas y sociales, hacen de la evaluación de los MMHH un tema espinoso que es fundamental abordar desde un punto de vista mucho más amplio que el de la evaluación económica convencional.
Esto, sumado a la heterogeneidad internacional de métodos utilizados para tomar las decisiones de autorización y fijación de precio, hacen cada vez más necesario que los decisores sanitarios cuenten con herramientas metodológicas adecuadas para guiar sus decisiones de planificación, priorización y asignación de recursos.
En las últimas décadas, las herramientas de decisión más utilizadas han sido la evaluación económica y el análisis de impacto presupuestario. Sin embargo,actualmente los decisores están contemplando cada vez más aspectos; factores clínicos y no clínicos que no son tomados en cuenta por estas herramientas, sobre todo en el ámbito de las enfermedades raras (EERR). Algunos de estos factores o criterios son la gravedad de la enfermedad, la disponibilidad o no de alternativas terapéuticas, el tamaño de la población afectada, los resultados reportados por los pacientes, la adherencia y persistencia al tratamiento, la calidad de la evidencia disponible, el grado de grado de innovación tecnológica o el valor para la salud pública1,2.
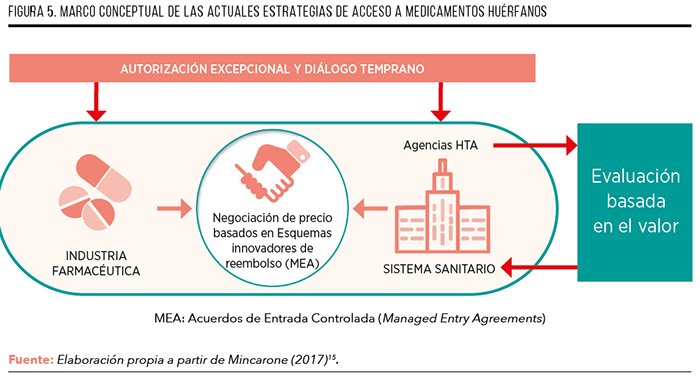
Sin embargo, el problema radica en que, por lo general, no suele explicitarse el tipo de criterios que considera el decisor y la importancia concreta que le da a cada uno de ellos. En efecto, a menudo, existe falta de transparencia en los criterios que determinan las decisiones de financiación, ya sea de manera consciente o subconsciente, por lo que, en algunos casos y sobre todo en el de los MMHH, el proceso de decisión se ha llegado a denominar “caja negra”3. En este contexto, el Análisis de Decisión Multi-Criterio (MCDA o MCDA por sus siglas en inglés, (Multi-Criteria Decision Analysis) se presenta como una solución que puede aportar congruencia y transparencia a la hora de decidir la financiación y el acceso a fármacos nuevos.
¿Y QUÉ ES EL MCDA?
En el debate sobre cómo conjugar innovación y sostenibilidad con las necesidades de los pacientes, aparece el MCDA. Se trata de una herramienta metodológica que ayuda a segmentar un problema o situación de forma explícita en términos de elección, ordenación o clasificación de alternativas para informar y facilitar la toma de decisiones, pero que en ningún caso reemplaza la toma de decisiones per se. La decisión final dependerá siempre de muchos factores, entre los que pueden figurar cuestiones políticas y juicios de valor, además de la evidencia clínica, económica y social.
En el ámbito de las enfermedades raras, los decisores están contemplando cada vez más aspectos; factores clínicos y no clínicos que no son tomados en cuenta por las herramientas tradicionales
Teniendo esto en cuenta, el MCDA puede ser muy útil como instrumento de ayuda a la toma de decisiones, bajo condiciones de correcta elección y aplicación de la técnica, ya que ayuda a incrementar la consistencia y transparencia de las decisiones. En efecto, el MCDA permite sistematizar el proceso de decisión en diferentes etapas, de manera estructurada e incorporando de forma explícita las preferencias de los agentes implicados a lo largo del proceso, tanto decisores como otros agentes relevantes. Esto aporta claridad sobre los criterios que son sustanciales a la hora de evaluar un fármaco y la importancia relativa de cada uno de estos criterios, lo cual resulta de mucha utilidad en contextos de alta complejidad, cómo es la evaluación de los MMHH.
Además de las decisiones de financiación de nuevos fármacos, en el sector sanitario los resultados del MCDA pueden usarse también para evaluar el beneficio-riesgo de diferentes alternativas, analizar una decisión de cartera, establecer decisiones de priorización de uso de recursos, evaluar una tecnología sanitaria o establecer criterios de priorización en el acceso de los pacientes a las innovaciones o a las prestaciones existentes4.
EL AUGE DEL MCDA EN EL SECTOR SANITARIO
Si bien es cierto que la aplicación del MCDA es relativamente reciente en el ámbito sanitario, este método ha sido utilizado ampliamente durante las últimas décadas tanto en el sector público como en el privado, como por ejemplo, en la toma de decisiones de transporte, energía, medio ambiente, defensa, inmigración o inversiones5.
En el sector sanitario, esta herramienta ha experimentado un auge en los últimos años, que se refleja en el creciente número de estudios publicados. Entre 2016 y 2018, las publicaciones de MCDA en el ámbito de las EERR superó al número de publicaciones totales que se habían realizado hasta ese año (Figura 1).
Entre las publicaciones sobre MCDA en MMHH cabe distinguir dos grupos diferenciados de estudios: modelos teóricos, mayormente con MMHH simulados; y otros estudios de aplicación práctica en condiciones de evaluación y datos reales.

EL MCDA FRENTE A LA EVALUACIÓN ECONÓMICA
Son muchos los autores que afirman que los métodos convencionales de evaluación de tecnologías sanitarias, incluida la evaluación económica, no permiten valorar la multiplicidad de dimensiones, criterios e implicaciones de índole ética, política, social y económica asociados a los MMHH7. Combinar adecuadamente todas las variables pertinentes en las decisiones de financiación exige intercambiar opiniones y deliberar sobre estas dimensiones. En el caso de los MMHH, la contraposición de criterios, valores y posturas se intensifican7.
En este contexto, el MCDA es uno de los instrumentos que puede ayudar a superar algunas de estas dificultades. Una de las ventajas que trae consigo frente a otros instrumentos es que dispone de una taxonomía de herramientas metodológicas para combinar o agregar múltiples criterios en un solo parámetro de valor7.
En este sentido, actualmente se debate sobre la utilidad del MCDA como una herramienta complementaria a la evaluación económica. En nuestra opinión, sería un error interpretar el MCDA como un sustituto del análisis coste-efectividad. Por el contrario, la combinación de múltiples criterios se plantea como un complemento de los métodos de evaluación convencionales, no como alternativa para sustituirlos.
Los métodos convencionales de evaluación de tecnologías sanitarias, incluida la evaluación económica, no permiten valorar
la multiplicidad de dimensiones, criterios e implicaciones
La implementación de una metodología de MCDA reflexiva para apoyar la toma de decisiones sobre MMHH estaría alineada con una de las nueve recomendaciones establecidas por el Grupo de Trabajo Europeo para la Evaluación del Valor y los Procesos de Financiación en Enfermedades Raras (ORPH-VAL)8, que propone que la evaluación de MMHH debería considerar todos los elementos relevantes del valor del fármaco, en un marco multidimensional apropiado, y trascendiendo los atributos incluidos en las evaluaciones convencionales.
Dadas las peculiaridades de los MMHH, a menudo no alcanzan los umbrales establecidos por el análisis coste-efectividad. Aunque la lógica del coste-efectividad es ampliamente reconocida como elemento necesario para guiar la toma de decisiones con recursos limitados, ninguna de las Agencias Evaluadoras de Tecnologías Sanitarias que tienen en cuenta la evaluación económica utilizan como única métrica la relación coste-efectividad incremental (RCEI). Para aquellos que no estén familiarizados con esta técnica, el RCEI implica priorizar las intervenciones o tecnologías sanitarias no sólo según su efectividad, sino según el balance entre sus costes y los resultados en salud comparados frente a su mejor alternativa. No obstante, el criterio de la efectividad/coste de oportunidad es una condición necesaria, pero no suficiente, para guiar la asignación de recursos sanitarios. Una de las posibles debilidades de la evaluación económica reside en que no permite tomar en cuenta las diferentes dimensiones de una tecnología sanitaria ni incorporar las preferencias individuales y sociales de los agentes que intervienen en el proceso, al igual que una de las debilidades del MCDA es que no incorpora de forma adecuada el concepto de coste de oportunidad, motivo por el que este tipo de abordajes son claramente complementarios y no excluyentes.
¿CÓMO REALIZAR UN MCDA?
Generalmente, la realización de todo MCDA comprende una serie de fases comunes, que conllevan la definición del problema, la selección y estructuración de los criterios de decisión, la recopilación de evidencia, la asignación de ponderaciones a los criterios, la evaluación de las alternativas a través de pesos o puntuaciones, el cálculo de los valores agregados, el manejo de la incertidumbre y la interpretación de resultados (Figura 2)5.

Se han publicado algunas guías de buenas prácticas que han examinado la aplicación del MCDA10. Una de las más relevantes ha sido publicada por el ISPOR Task Force Report11. Entre algunos ejemplos de buenas prácticas en el desarrollo y aplicación de un MCDA, se encuentran la realización de pruebas piloto para garantizar el tipo de método de ponderación y puntuación; conocer y entender la heterogeneidad de los distintos tipos de agentes (stakeholders) que formarán parte del proceso de evaluación y manejar el tipo de incertidumbre asociado al modelo.
CRITERIOS A CONTEMPLAR EN UN MCDA EN EERR
La elección de los criterios es un paso fundamental en todo MCDA, que puede variar en función del objetivo planteado. Los criterios incluidos deben cumplir algunas condiciones imprescindibles: ser no redundantes, independientes, completos, operacionales y medibles. Cada criterio debe contribuir al resultado o beneficio con independencia de los demás y evitando la duplicidad.
Cuando se analizan los criterios empleados en los MCDA publicados en la literatura, existe un alto grado de coincidencia en cuanto a los más utilizados en la evaluación de MMHH. En general, los aspectos clínicos (impacto en la salud, gravedad de la enfermedad y necesidades terapéuticas no cubiertas) son los criterios más habituales, mientras que los aspectos económicos (ya sea impacto presupuestario o coste-efectividad) juegan un papel minoritario y se incluyen en menos de la mitad de los MCDA. Es decir, no existe un consenso sobre el grado de importancia de su inclusión y a menudo se incurre en una definición equívoca de los criterios, solapamiento y doble contabilización de costes7.
La selección de los criterios puede realizarse de dos formas alternativas. Una primera es ad hoc para una decisión en concreto, empleando un método top-down en donde los criterios se seleccionan antes de conocer las alternativas a evaluar o un método bottom-up, en donde las alternativas son seleccionadas primero y los criterios son dependientes de éstas. La otra forma es empleando un marco general de criterios predefinidos.
Uno de los marcos más conocidos y utilizados en el sector salud es el marco EVIDEM (Evidence and Value: Impact on Decision-Making), diseñado inicialmente en 2006, y que ha sido adaptado en diversas ocasiones12. Actualmente, el EVIDEM cuenta con una serie de criterios genéricos y universalmente operacionales (concretamente, 13 criterios cuantitativos agrupados en 5 dominios) y 7 criterios contextuales que se distribuyen en dos grupos de dominios.
El MCDA es una herramienta metodológica que ayuda a segmentar un problema o situación de forma explícita
También existen marcos específicos ad hoc creados en el ámbito de las EERR. Uno de los primeros fue el desarrollado por Hughes-Wilson en 2012, con la intención de servir como herramienta de evaluación en la fijación de precio y reembolso en el contexto de la Unión Europea. Los criterios propuestos fueron la rareza, gravedad de la enfermedad, disponibilidad de otras alternativas (necesidades terapéuticas no cubiertas), nivel de impacto sobre la condición que ofrece el nuevo tratamiento, si el medicamento puede usarse en una o más indicaciones, el nivel de investigación llevado a cabo por el laboratorio, junto con otros factores, como la complejidad de fabricación y las medidas de seguimiento requeridas por las autoridades13.
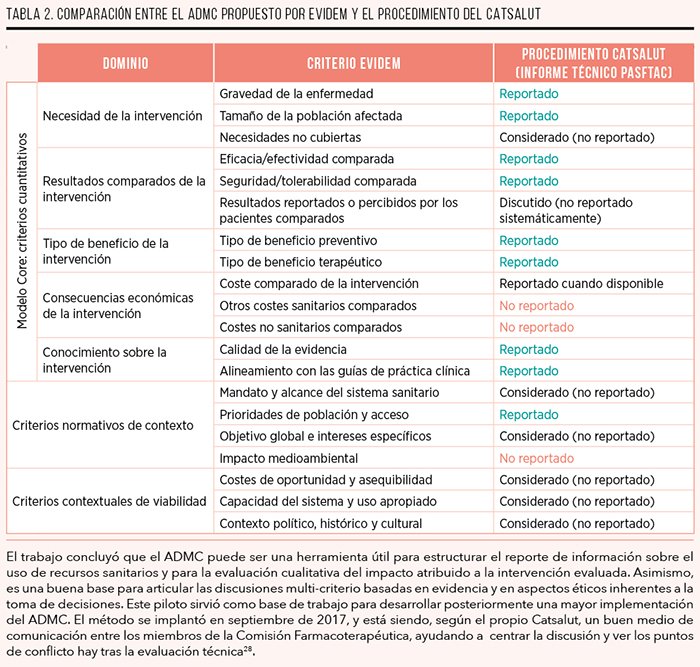
En cuanto a la adaptación de los marcos predefinidos en España, cabe destacar la experiencia piloto de Cataluña en 2015, a través de la cual se comparó el marco EVIDEM con los criterios usados por el Comité Fármaco-terapéutico del CatSalut (PASFTAC) para evaluar los MMHH, concluyendo que eran similares y que el MCDA podía ser una herramienta útil complementaria de sus métodos de evaluación14.
Otra adaptación, más reciente, del marco EVIDEM en el ámbito de los MMHH realizada en el contexto español es la publicada por Badía et al. en 2019, donde los autores proponen un marco reflexivo con atributos relevantes que faciliten la toma de decisionesl15. Para ello, un grupo multidisciplinar de expertos en EERR en España, formado por representantes del Grupo de Enfermedades Raras y Medicamentos Huérfanos de la SEFH (OrPhar-SEFH), la Federación Española de Enfermedades Raras(FEDER), el Instituto de Salud Carlos III, el Instituto de Investigación en Enfermedades Raras (IIER) y el Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), entre otros, adaptaron el marco EVIDEM, proponiendo modificaciones a la definición y/o escalas de los criterios (Tabla 1). La validación de los criterios se realizó en una segunda fase, probando posteriormente el marco en dos medicamentos huérfanos concretos.

TÉCNICAS DE PONDERACIÓN, PUNTUACIÓN, MODELIZACIÓN Y MANEJO DE LA INCERTIDUMBRE EN EL MCDA
En la fase de modelización, la evidencia recogida es objeto de cuantificación por parte de los evaluadores para identificar la mejor alternativa, incorporando ponderaciones y puntuaciones explícitas.
El objetivo de la ponderación de las alternativas es capturar las preferencias y prioridades de los agentes implicados sobre la importancia que tiene para ellos cada uno de los criterios cuantitativos, es decir, cuanto de importante es el criterio “x” frente al “z”. Estas ponderaciones serán probablemente distintas para cada grupo de actores. La selección del método de ponderación dependerá de las características del problema de decisión.
Por su parte, el objetivo de la puntuación es valorar de manera cuantitativa las intervenciones evaluadas según los diferentes criterios que se han considerado previamente relevantes (en la fase de ponderación). La selección del método de puntuación debe hacerse teniendo en cuenta la carga cognitiva de los actores, el nivel de precisión requerido, los fundamentos teóricos y la heterogeneidad de los agentes.
Los métodos de ponderación más utilizados en los MCDA de MMHH son el método jerárquico (escala 1 a 5) y el método direc-to (reparto de 100 puntos). Sin embargo, algunos estudios han utilizado métodos de elección discreta. Un ejemplo de ello es el estudio resultante de un proyecto europeo (López-Bastida et. al 2019)16, que llevó a cabo un piloto evaluando la viabilidad de utilizar el método de elección discreta para investigar las preferencias individuales desde la perspectiva del pagador en relación a la financiación de MMHH. El estudio, aplicado en Inglaterra, Francia, Alemania, Italia y España, reveló una preferencia relativa por algunos atributos sobre otros: el coste del tratamiento, la mejora de la salud, la relación calidad-precio (value for money) y las alternativas disponibles recibieron la mayor importancia. La gravedad de la enfermedad, la población a la cual va dirigida la terapia, los tiempos de espera y los efectos secundarios también son valores sociales importantes que no deben ignorarse.
EXISTEN DOS GRANDES GRUPOS DE MÉTODOS DE PONDERACIÓN Y PUNTUACIÓN:
- Los métodos de composición, que incluyen los métodos directos, métodos jerárquicos y métodos de concordancia;
- Los métodos de des composición,que incluyen los modelos de elección discreta.

La forma en la que se construyen los modelos de agregación de preferencias también es un elemento que diferencia a los distintos MCDA. La valoración puede hacerse de distintas maneras: directamente mediante un valor cuantitativo; comparando alternativas; o comparando escenarios que incluyan distintas combinaciones de criterios y valoraciones. Así, podemos distinguir entre modelos de medida del valor, modelos outranking y modelos por objetivos o niveles de referencia9.
Como en todos los ejercicios de modelización, el resultado depende de las asunciones realizadas y de las decisiones tomadas a lo largo de la construcción del modelo, no solo de los inputs y de la propia estructura del mismo.
Un elemento a tener muy en cuenta es la incertidumbre estructural asociada al diseño del modelo construido, sin olvidar que existen otros tipos de incertidumbre, como la estocástica o la relacionada con la evidencia clínica. Parte de esta incertidumbre se debe manejar a través del análisis de sensibilidad, que proporciona una medida de validez de los resultados.
LOS STAKEHOLDERS
Uno de los puntos más relevantes del MCDA es que la herramienta en sí misma se fundamenta en un proceso deliberativo, explícito y de ordenación de preferencias de los agentes implicados en el proceso de toma de decisiones. La composición y el perfil de cada uno de los miembros del comité repercutirá por tanto en los resultados del MCDA. De ahí la importancia del proceso de selección de los evaluadores, ya que determinará la validez interna y externa de los resultados, debiendo detallarse cómo y por qué se han elegido y explicitar posibles conflictos de interés. La multidisciplinariedad es un elemento fundamental para dar voz a todas las partes involucradas en la decisión o afectadas por la misma y que enriquecerá el intercambio de valoraciones individuales y el análisis en su conjunto.
Según una revisión de algunos MCDA publicados en MMHH, los comités multidisciplinares estaban generalmente formados por entre 8 y 28 miembros. Los actores más comúnmente representados fueron los clínicos, presentes en todos los comités, seguidos de los pacientes o representantes de asociaciones de pacientes (en el 90% de los comités) y decisores/pagadores (60%). El resto de agentes fueron menos frecuentes (economistas de la salud, farmacéuticos, representantes de la industria farmacéutica, enfermería, etc), siendo la composición de los comités sustancialmente variable entre los distintos estudios17.
Las experiencias de aplicación real a nivel de organismos evaluadores y pagadores todavía son escasas, aunque ya se han producido algunos avances a nivel tanto nacional como internacional
Como era de esperar, se observan diferencias entre las preferencias y prioridades de los distintos actores. En general, los pacientes reparten sus ponderaciones de forma más igualitaria, dando más peso que el resto a la afectación de la vida cotidiana. Por su parte, los clínicos dan más peso a la gravedad de la patología y a la efectividad del tratamiento, mientras que los pagadores ponderan en mayor medida los costes y la falta de alternativas terapéuticas.

APLICABILIDAD DEL MCDA EN LA EVALUACIÓN DE MMHH
El creciente número de estudios publicados recientemente revela un mayor interés por utilizar el MCDA en EERR. Sin embargo, la mayoría son aplicaciones teóricas o ejercicios aislados de evaluación de fármacos concretos, impulsados por la industria farmacéutica, por ejemplo en hipertensión arterial pulmonar18, carcinoma de tiroides diferenciado refractario al yodo radioactivo19, tumores neuroendocrinos gastroenteropancreático20, linfoma no-Hodgkin21, síndrome de Turner22 o inmunodeficiencia23.
Las experiencias de aplicación real a nivel de organismos evaluadores y pagadores todavía son escasas, aunque ya se han producido algunos avances a nivel tanto nacional como internacional.
En el contexto nacional, podemos resaltar algunos pilotos desarrollados a nivel regional. Si bien no todos se refieren específicamente al ámbito de los MMHH, sirven de inicio y exploración para poder establecer consensos sobre el mejor uso y aplicabilidad de esta herramienta. Como ya hemos mencionado, Cataluña fue pionera en este sentido, poniendo en marcha unpiloto en el PASFTAC para testar cómo de alejado era su proceso del que se derivaría de un MCDA más formal14.
También se han realizado otros ejercicios conceptuales, contando con decisores y representantes de organismos relevantes, para desarrollar un marco específico de evaluación de MMHH a nivel nacional15, o para conocer las preferencias de los decisores en cuanto a los criterios de evaluación de los fármacos. Por ejemplo, se llevó a cabo un proyecto para analizar las preferencias de un grupo de evaluadores de la Agencia Española de Medicamentos y Productos Sanitarios, que sirvió para poner de manifiesto que algunos de los criterios del marco EVIDEM no se contemplan explícitamente en los Informes de Posicionamiento Terapéutico, como es el caso de la gravedad de la enfermedad, la calidad de la evidencia y las necesidades no cubiertas24. Otro trabajo analizó las preferencias de evaluadores de las comisiones regionales de Andalucía, País Vasco y Cataluña respecto a los distintos criterios, siendo los más valorados la gravedad de la enfermedad, la eficacia y seguridad comparada, la calidad de laevidencia y el tipo de beneficio terapéutico25.
A conclusiones similares llegó un trabajo publicado más recientemente, realizado a partir de un total de 98 evaluadores y deci-sores (87% farmacia hospitalaria, 10% clínicos, 1% pacientes y 1% economistas de la salud), según los cuales los criterios más importantes son la gravedad, la eficacia y la calidad de la evidencia, y los menos relevantes los costes no sanitarios, el alineamiento con las guías de práctica clínica y el tamaño de la población afectada (Figura 4)26.

A nivel de microgestión, algunos hospitales, como el Virgen de la Macarena de Sevilla, han utilizado el MCDA para evaluar nuevos medicamentos en su comisión farmacoterapéutica, demostrando que la herramienta era útil para detallar los criterios considerados y entender sus procesos de evaluación en la incorporación de nuevos fármacos27.
Cabe también señalar el creciente interés que han demostrado algunas agencias regionales de evaluación de tecnologías sanitarias en el desarrollo metodológico del MCDA. Este es el caso de la agencia de Andalucía, que desarrolló una guía para la elaboración de recomendaciones basadas en MCDA y de la agencia del País Vasco, que desarrolló un marco contextual multi-criterio recopilado en 26 criterios cualitativos de dominios éticos, organizativos, legales, sociales y medioambientales, entre otros28,29.
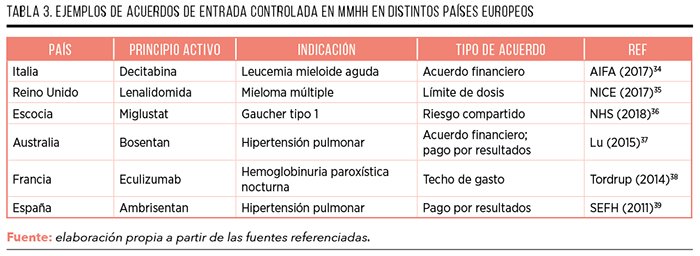
A nivel internacional, también se observa un uso creciente del MCDA en la práctica real, con múltiples ejemplos de utilización en distintos países, sobre todo en la priorización de intervenciones de alto impacto y MMHH financiados de manera pública (Tabla 2)30.

Otros ejemplos los encontramos en países como Bélgica, en donde se propuso un marco de referencia para mejorar la rendición de cuentas en el marco de reembolso de prestaciones públicas sanitarias, Colombia, donde la Comisión de Regulación enSalud utilizó el marco EVIDEM para actualizar las prestaciones del Plan Obligatorio de Salud4 o Lombardía (en Italia), que ha utilizado un enfoque MCDA reflexivo combinado con el modelo central de la Red Europea para la Evaluación de Tecnologías Sanitarias para evaluar y tomar decisiones de cobertura y reembolso de medicamentos de alto impacto desde 201231.

LAS DOS CARAS DEL MCDA
El MCDA cuenta con una serie de ventajas, entre las que destaca el hecho de que permite analizar las intervenciones desde una visión holística, considerando explícitamente atributos de valor que pueden ser relevantes para la sociedad, que van más allá de los tradicionales de eficacia, seguridad y precio. Además, entre los atributos a considerar se pueden incluir elementos cualitativos y contextuales.
Al ser el MCDA una herramienta que ayuda a formalizar el proceso de decisión y armoniza el trabajo conjunto de todos los agentes que intervienen durante el mismo, permite dotar de mayor transparencia y rendición de cuentas a la toma de decisiones, puntos muy necesarios en contextos opacos y complejos como la evaluación de medicamentos huérfanos. Además, permite el debate reflexivo, argumentado y multidisciplinar, ayudando a poner en común y entender las distintas perspectivas, incluyendo la del paciente.
Sin embargo, el MCDA también adolece de ciertas limitaciones. Entre ellas, cabe citar el riesgo de doble contabilización de algunos criterios, la omisión de otros aspectos relevantes, como el coste de oportunidad, la subjetividad inherente al grupo que realiza la evaluación, el potencial uso sesgado de la herramienta, la falta de comparabilidad entre distintos ejercicios o la difícil generalización de los resultados4. En el caso de las enfermedades raras, otro punto débil del MCDA es que en algunas ocasiones puede ser difícil la elección de un comparador adecuado, dado que se trata de enfermedades para la que no existen alternativas terapéuticas, lo que hace que el resultado no esté acorde a la estrategia de posicionamiento del fármaco.
EN BUSCA DEL VALOR DEL FÁRMACO HUÉRFANO
En conclusión, el MCDA es una herramienta metodológica de creciente uso para evaluar los MMHH, ya que permite capturar elementos de valor adicionales al análisis coste-efectividad estándar, desde una visión más amplia y holística que puede respaldar diferentes tipos de decisiones en el ámbito sanitario. Aún así, hace falta mayor consenso y homogenización en su aplicación práctica.
En todo caso, no debe olvidarse que el MCDA no puede sustituir a la toma de decisiones en sí misma, ni ser la única herramienta empleada en el proceso de evaluación, sino que debería utilizarse como un instrumento más de apoyo a la toma de decisiones.
REFERENCIAS
- Golan O, Hansen P, Kaplan G, Tal O. Health technology prioritization: Which criteria for prioritizing new technologies and what are their relative weights? Health Policy. 2011;102(2-3):126-35, doi: 10.1016/j.healthpol.2010.10.012.
- Baltussen R, Niessen L. Priority setting of health interventions: the need for multi-criteria decision analysis. Cost effectiveness and resource allocation. 2006;4(1):14.
- Picavet E, Morel T, Cassiman D, Simoens S. Shining a light in the black box of orphan drug pricing. Orphanet journal of rare diseases. 2014;9(1):62.
- Zozaya González, N, Oliva Moreno, J, Hidalgo Vega, A, Alcalá Revilla, B, García Ruíz, A, García-Agua Soler, N, et al. El análisis de decisión multi-criterio en el ámbito sanitario. Fundación Weber; 2018.
- Thokala P, Devlin N, Marsh K, Baltussen R, Boysen M, Kalo Z, et al. Multiple Criteria Decision Analysis for Health Care Decision Making—An Introduction: Report 1 of the ISPOR MCDA Emerging Good Practices Task Force. Value in Health. 2016;19(1):1-13, doi: 10.1016/j.jval.2015.12.003.
- Baran-Kooiker A, Czech M, Kooiker C. Multi-Criteria Decision Analysis (MCDA) Models in Health Technology Assessment of Orphan Drugs—a Systematic Literature Review. Next Steps in Methodology Development? Front Public Health. 2018;6, doi: 10.3389/fpubh.2018.00287.
- Campillo-Artero, Carlos. Aplicación del análisis de decisión multi-criterio a los medicamentos huérfanos. Capítulo 8 en el libro: El análisis de decisión multi-criterio en el ámbito sanitario. Madrid: Fundación Weber; 2018.
- Annemans L, Aymé S, Le Cam Y, Facey K, Gunther P, Nicod E, et al. Recommendations from the European Working Group for Value Assessment and Funding Processes in Rare Diseases (ORPH-VAL). Orphanet J Rare Dis. 2017;12(1):50, doi: 10.1186/s13023-017-0601-9.
- Thokala P, Duenas A. Multiple Criteria Decision Analysis for Health Technology Assessment. Value in Health. 2012;15(8):1172-81, doi: 10.1016/j.jval.2012.06.015.
- Trapero-Bertran, M. Revisión de guías metodológicas y de buenas prácticas en MCDA. Capítulo 6 en el libro: El Análisis de Decisión Multi-Criterio en el ámbito sanitario. Fundación Weber; s. f.
- Marsh K, IJzerman M, Thokala P, Baltussen R, Boysen M, Kaló Z, et al. Multiple Criteria Decision Analysis for Health Care Decision Making—Emerging Good Practices: Report 2 of the ISPOR MCDA Emerging Good Practices Task Force. Value in Health. 2016;19(2):125-37, doi: 10.1016/j.jval.2015.12.016.
- EVIDEM Collaboration. EVIDEM Framework 10 th edition – Overview. [accedido 4 enero 2018]. Disponible en: https://www.evidem.org/evidem-framework/.
- Hughes-Wilson W, Palma A, Schuurman A, Simoens S. Paying for the Orphan Drug System: break or bend? Is it time for a new evaluation system for payers in Europe to take account of new rare disease treatments? Orphanet journal of rare diseases. 2012;7(1):74.
- Gilabert-Perramon A, Torrent-Farnell J, Catalan A, Prat A, Fontanet M, Puig-Peiró R, et al. Drug evaluation and decision making in Catalonia: development and validation of a methodological framework based on multi-criteria decision analysis (MCDA) for orphan drugs. International Journal of Technology Assessment in Health Care. 2017:1-10, doi: 10.1017/S0266462317000149.
- Badia X, Chugani D, Abad MR, Arias P, Guillén-Navarro E, Jarque I, et al. Development and validation of an MCDA framework for evaluation and decision-making of orphan drugs in Spain. Expert Opinion on Orphan Drugs. 2019;7(7-8):363-72, doi: 10.1080/21678707.2019.1652163.
- López-Bastida J, Ramos-Goñi JM, Aranda-Reneo I, Trapero-Bertran M, Kanavos P, Rodriguez Martin B. Using a stated preference discrete choice experiment to assess societal value from the perspective of decision-makers in Europe. Does it work for rare diseases? Health Policy. 2019;123(2):152-8, doi: 10.1016/j.healthpol.2018.11.015.
- Zozaya, N, Galindo, J, Alcalá, B, Hidalgo-Vega, A. El análisis de decisión multi-criterio como herramienta para la toma de decisiones en medicamentos huérfanos: una revisión de la literatura. IX Congreso Internacional de Medicamentos Huérfanos y Enfermedades Raras, Sevilla; 2019.
- Jimenez A, Ais A, Acuña L, González M, Paco N, Gil A. Determining The Value of Selexipag For The Treatment of Pulmonary Arterial Hypertension (PAH) In Spain By Multi-Criteria Decision Analysis (MCDA). Value in Health. 2017;20(9):A570, doi: 10.1016/j.jval.2017.08.971.
- Wagner M, Khoury H, Bennetts L, Berto P, Ehreth J, Badia X, et al. Appraising the holistic value of Lenvatinib for radio-iodine refractory differentiated thyroid cancer: A multi-country study applying pragmatic MCDA. BMC Cancer. 2017;17, doi: 10.1186/s12885-017-3258-9.
- Wagner M, Samaha D, Cuervo J, Patel H, Martinez M, O’Neil WM, et al. Applying Reflective Multicriteria Decision Analysis (MCDA) to Patient–Clinician Shared Decision-Making on the Management of Gastroenteropancreatic Neuroendocrine Tumors (GEP-NET) in the Spanish Context. Adv Ther. 2018;35(8):1215-31, doi: 10.1007/s12325-018-0745-6.
- Garau M, Devlin NJ. Using MCDA as a Decision Aid in Health Technology Appraisal for Coverage Decisions: Opportunities, Challenges and Unresolved Questions. Multi-Criteria Decision Analysis to Support Healthcare Decisions. Springer, Cham; 2017. p. 277-98.
- Goetghebeur MM, Wagner M, Khoury H, Rindress D, Grégoire J-P, Deal C. Combining multicriteria decision analysis, ethics and health technology assessment: applying the EVIDEM decisionmaking framework to growth hormone for Turner syndrome patients. Cost Effectiveness and Resource Allocation. 2010;8(1):4.
- Sussex J, Rollet P, Garau M, Schmitt C, Kent A, Hutchings A. Multi-criteria decision analysis to value orphan medicines. 2013.
- Hernández C, Blázquez A, Gil A, Badia X. Relative Value of Evidem Mcda Framework for Reflective Drug Evaluation Among Therapeutic Positioning Report Evaluators From The Spanish Agency of Medicines. Value in Health. 2017;20(9):A699, doi: 10.1016/j.jval.2017.08.1806.
- Gilabert-Perramon A, Lens C, Betolaza JI, March JC, Espín J, Merino-Montero S, et al. Multi-Criteria Decision Analysis (MCDA): Common Tools for Different Needs Supporting Healthcare Decision Making in Spain. Value in Health. 2016;19(7):A489-90, doi: 10.1016/j.jval.2016.09.827.
- Badia X, Gil A, Shepherd J. PHP169 – MCDA EVIDEM Reference Value Framework for drug evaluation and decision making in Spain. Value in Health. 2018;21:S179, doi: 10.1016/j.jval.2018.09.1063.
- Roldán ÚB, Badia X, Marcos-Rodríguez JA, de la Cruz-Merino L, Gómez-González J, Melcón-de Dios A, et al. Multi-criteria decision analysis as a decision-support tool for drug evaluation: a pilot study in a pharmacy and therapeutics committee setting. Int J Technol Assess Health Care. 2018;34(5):519-26, doi: 10.1017/ S0266462318000569.
- Agencia de Evaluación de Tecnologías, Sanitarias de Andalucía. Grupo de trabajo de la guía para la elaboración de recomendaciones y criterios de uso adecuado. Guía para la elaboración de recomendaciones y criterios de uso adecuado de tecnologías sanitarias. 2017.
- Bayón Yusta, JC, Gutiérrez Iglesias, A, Galnares-Cordero, L, Gutiérrez-Ibarluzea, I. Síntesis de información relevante de apoyo a los MCDA (análisis de decisión multicriterio) para la toma de decisiones: proyecto metodológico. Informes de Evaluación de Tecnologías Sanitarias: OSTEBA.; 2019.
- Drake JI, de Hart JCT, Monleón C, Toro W, Valentim J. Utilization of multiple-criteria decision analysis (MCDA) to support healthcare decision-making FIFARMA, 2016. J Mark Access Health Policy. 2017;5(1), doi: 10.1080/20016689.2017.1360545.
- Radaelli G, Lettieri E, Masella C, Merlino L, Strada A, Tringali M. Implementation of Eunethta Core Model® in Lombardia: The Vts Framework. International Journal of Technology Assessment in Health Care; Cambridge. 2014;30(1):105-12, doi: http://dx.doi.org/10.1017/S0266462313000639.