Coral González, Jorge Barrios y Carlos Dévora
Departamentos de Health Economics & Public Affairs, Data & Technology y Health Economics & Market Access de Weber
INTRODUCCIÓN
Contexto actual del uso del Big Data en salud
La explosión de datos en la que estamos inmersos desde hace unos años nos ha conducido hacia el paradigma del Big Data, caracterizado por un tamaño que excede la capacidad de las herramientas de software convencionales para capturar, almacenar y analizar la información1,2. Gartner propuso en 2012 la definición de las “3V” del Big Data: gran volumen, velocidad y variedad de activos de información3. Las nuevas definiciones añaden también una cuarta dimensión, cada vez más relevante: la veracidad de estos datos4. Esta última “V” cobra especial importancia en el ámbito sanitario, con cuestiones como: ¿Se han registrado correctamente los diagnósticos, tratamientos, prescripciones, procedimientos y/o resultados?
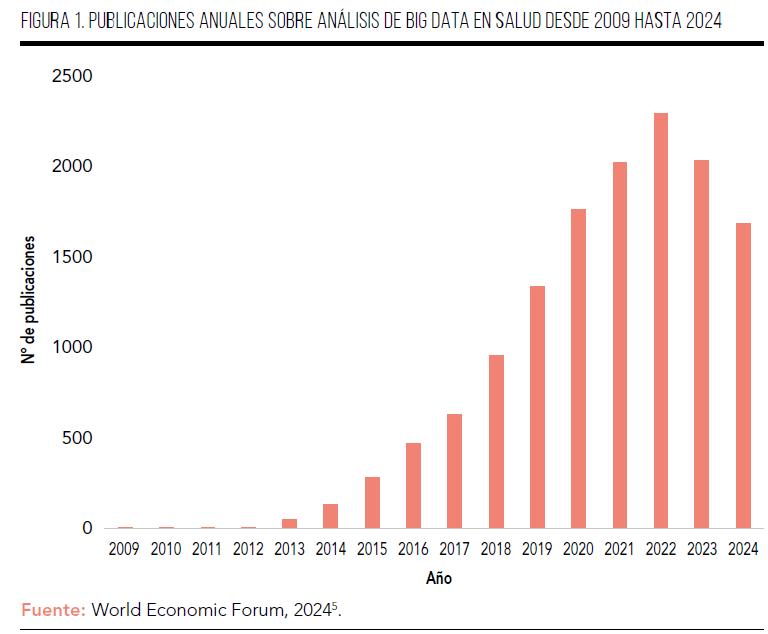
Los avances en la eficacia y seguridad de los medicamentos, la precisión de los diagnósticos y la orientación terapéutica más precisa dependen de tener estos datos de alta calidad. Evidencia de ello es el crecimiento exponencial de investigaciones sobre Big Data y Salud en los últimos años1,5 (Figura 1). Según datos del World Economic Forum, en el año 2020 se generaron a nivel global ~2.300 exabytes de datos en el sector sanitario6.
Dada la magnitud de estas cifras, en este artículo pretendemos explorar cómo los espacios y plataformas de datos impulsan la investigación y la evaluación económica en la salud y especialmente en las enfermedades raras (EERR), destacando las principales iniciativas nacionales y europeas. Asimismo, analizaremos el papel del Big Data y la interoperabilidad en el avance del diagnóstico, el pronóstico y los modelos predictivos, así como su aportación a la evaluación económica y a la toma de decisiones. También tendremos en cuenta los principales desafíos técnicos, legales y éticos, junto con las oportunidades que se abren para las EERR y el sistema sanitario en general.
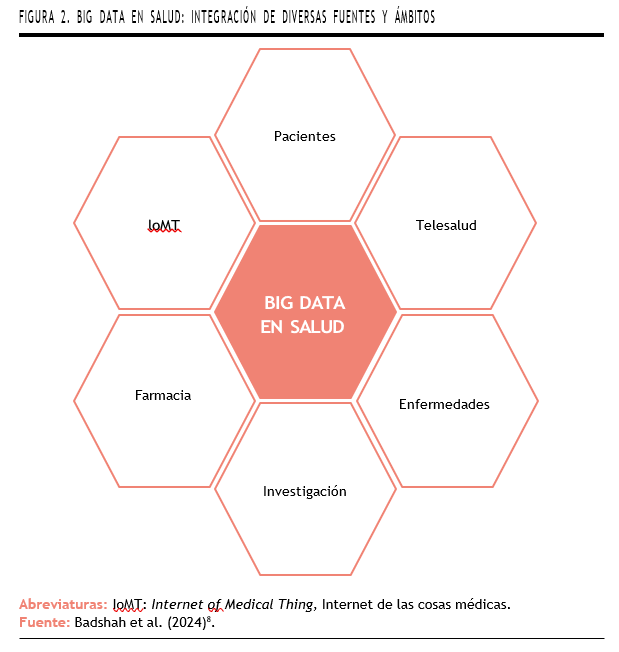
Hay que tener en cuenta que todos estos datos se recopilan a través de diversas fuentes, desde registros electrónicos, datos farmacéuticos y genómicos, ensayos clínicos o telemedicina, cuya implementación ha sido creciente desde la pandemia de la COVID-19. Otra fuente emergente de información es la conocida como el Internet de las Cosas Médicas (IOMT del inglés Internet of Medical Things)7, entre las que se encuentran las aplicaciones móviles, smartwatches y otro tipo de sensores (Figura 2)1.
A nivel internacional, la Organización Mundial de la Salud (OMS) anunció en 2024 el lanzamiento del programa “Impact Training for Big Data in Health Care” en Europa, enfocado en mejorar las capacidades de los países en torno al uso del Big Data en la salud9. Asimismo, durante el mes de octubre de 2025, la Comisión Europea ha publicado las convocatorias de proyectos de fondos de la Unión Europea (UE), de los cuales 22,5 millones de euros están destinados a apoyar la investigación biomédica y la implantación de la asistencia sanitaria personalizada a través de la Infraestructura Europea de Datos Genómicos; mientras que 14,4 millones de euros se destinarán al despliegue de soluciones basadas en la inteligencia artificial (IA) en el procesamiento de imágenes médicas10.
En el contexto español, el uso del Big Data y la IA también se está consolidando en el ámbito sanitario mediante diversas iniciativas institucionales. Ejemplo de ello es el equipo de evaluación de la Agència de Qualitat i Avaluació Sanitàries de Catalunya (AQuAS), que elaboró en 2024 la “Guía de evaluación de tecnologías de salud digital que incorporan inteligencia artificial”, donde propone los dominios fundamentales a evaluar para la correcta toma de decisiones informada11. Otro ejemplo lo encontramos en la herramienta “Analítica Farmacia”, desarrollada por el Ministerio de Sanidad, que emplea IA para generar modelos predictivos en los procedimientos de precio y reembolso de los medicamentos12. Aplicar estas tecnologías que combinan Big Data e IA al ámbito de la salud ofrece numerosas ventajas, algunas de las cuales aparecen recogidas en la Tabla 1.
TABLA 1. VENTAJAS DEL USO DEL BIG DATA EN LA SALUD
| Área |
Ventajas |
| Optimización del manejo clínico |
Permite realizar investigaciones comparativas de coste-efectividad, identificando los tratamientos y diagnósticos más eficientes, mejorando la asignación de recursos y la toma de decisiones médicas. |
| Impulso a la investigación y desarrollo (I+D) |
Facilita el uso de modelos predictivos para optimizar la selección de pacientes candidatos y acelerar el desarrollo de nuevos fármacos.
Mejora el diseño de ensayos clínicos y la selección de pacientes, aumentando las tasas de éxito y reduciendo los tiempos de llegada de los tratamientos al mercado. |
| Medicina personalizada |
Hace posible ejecutar secuenciaciones genéticas más rápidas y económicas, integrando los resultados en la historia clínica del paciente.
Facilita la predicción de riesgos de enfermedad o reingreso hospitalario. |
| Monitorización y seguimiento remoto |
Facilita la recolección y análisis en tiempo real de datos procedentes de dispositivos médicos hospitalarios o domiciliarios, ayudando a predecir eventos adversos y mejorar la seguridad del paciente. |
| Prevención del fraude y mejora administrativa |
Permite analizar rápidamente millones de solicitudes de reembolso para detectar fraudes, abusos o gastos innecesarios en los sistemas de salud. |
Fuente: Adaptado de Raghupathi et al.13
Retos específicos en las EERR
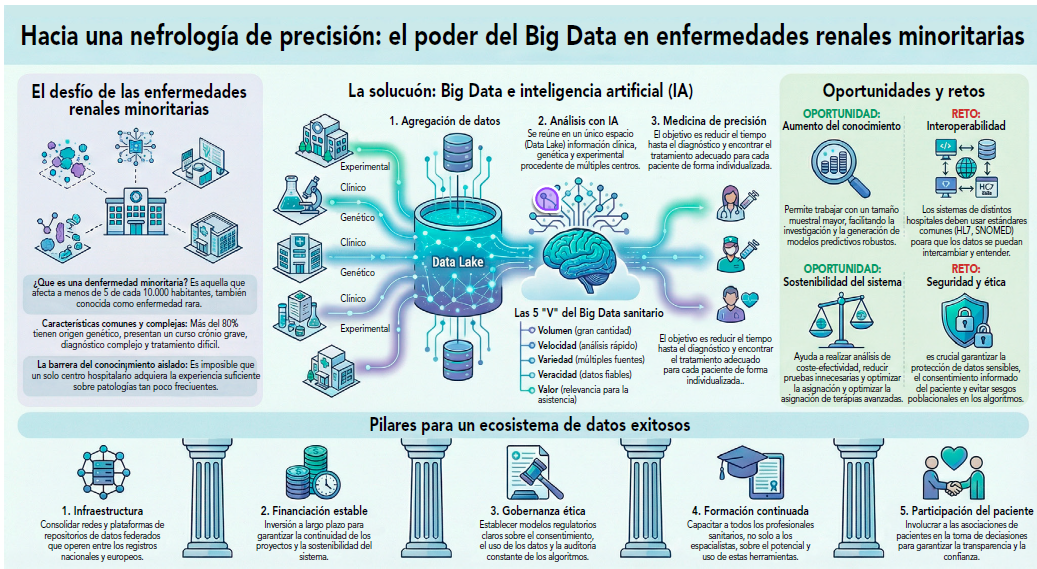
Dentro del ámbito de la salud, uno de los campos que más podrían beneficiarse de la implementación del Big Data y el uso de espacios de datos compartidos es el de las enfermedades raras (EERR). Las personas con EERR suelen enfrentarse a una verdadera “odisea diagnóstica” que, en los países desarrollados, conlleva un promedio de 5 a 6 años.14. Además, sólo alrededor del 5% de las EERR cuenta tratamientos específicos efectivos15, y los medicamentos huérfanos (MMHH) siguen siendo escasos, costosos y con una accesibilidad aún limitada16. En esencia, los dos problemas derivan, principalmente, de una misma causa: la limitada disponibilidad de datos y la fragmentación del conocimiento generado en este ámbito, además de la necesidad de las empresas de recuperar su inversión a través de la venta de sus productos, lo que se complica dado el número reducido de pacientes.
La escasez de datos se debe, principalmente, a la falta de personal especializado y de infraestructuras de investigación adecuadas, como registros de pacientes o cohortes de estudios clínicos suficientemente amplias, así como de biobancos que dispongan de la información necesaria17, a lo que se suma la fragmentación de la poca evidencia disponible y la incertidumbre asociada a esta evidencia. La ausencia de guías de práctica clínica que garanticen el manejo adecuado de los pacientes y la falta de integración de los datos entre las distintas especialidades médicas por las que circulan durante su recorrido asistencial los pacientes de EERR hace que los datos sean heterogéneos y estén dispersos18–20. Estas limitaciones ponen de manifiesto la importancia de desarrollar nuevas herramientas y enfoques que permitan superar estas brechas y avanzar de forma coordinada, siendo el Big Data y el uso sistemático de datos clínicos y de investigación una vía prometedora para paliar tanto la escasez como la fragmentación actuales.
Como comentamos al principio, este artículo analizará cómo los espacios y plataformas de datos pueden transformar la investigación y la evaluación económica en las EERR, revisando las principales iniciativas nacionales y europeas y el papel del Big Data y la interoperabilidad en la mejora del diagnóstico, pronóstico y tratamiento mediante modelos predictivos.
ESPACIOS DE DATOS EN SALUD Y SU IMPACTO EN LAS EERR
Concepto de espacio de datos y ecosistema federados
Según la Agencia Española de Protección de Datos (AEPD), un espacio de datos es «una infraestructura federada y abierta para permitir el acceso soberano de datos, basada en una gobernanza, políticas, reglas y estándares que definen un marco de confianza para todos los intervinientes»21. Los espacios de datos convierten la información en un activo estratégico que facilita el uso de herramientas como el Big Data, el aprendizaje automático y la automatización de procesos, entre otros22. En contextos sanitarios, este uso de los datos conduce a soluciones más eficientes y adaptadas a las necesidades reales de las personas, supliendo la falta de datos unificados, los registros médicos incompletos y la escasez de ensayos clínicos.
Vinculados a estos espacios se encuentran los ecosistemas de datos federados, compuestos por una serie de nodos descentralizados e interconectados que permiten reutilizar los datos sin necesidad de transferirlos desde los proveedores hacia los usuarios23,24. Una de las aplicaciones más potentes de estos ecosistemas es el aprendizaje automático federado (federated learning), una técnica de IA que permite entrenar modelos sin centralizar la información.
Gracias a estas redes, hospitales o laboratorios pueden compartir algoritmos y realizar consultas sobre sus datos de manera colaborativa, manteniendo la información sensible local y segura. En este proceso, los algoritmos se ejecutan directamente sobre los datos locales y únicamente se comparten los resultados o los ajustes del modelo, nunca los datos originales. Este enfoque combina la colaboración científica con la protección de la privacidad, facilitando el análisis de grandes volúmenes de información distribuidos en múltiples instituciones23,25.
Algunos ejemplos de aplicaciones clínicas de ecosistemas de datos federados de salud ya existentes las encontramos sobre todo en áreas como la oncología, (ej. The Federated Tumor Segmentation (FeTS) initiative26, formada por 30 institutos de atención médica que trabajan para sortear las barreras de detección tumoral) o la COVID-19 (formada por una red de 12 hospitales británicos27).
En el contexto de las EERR, este modelo reduce las barreras para el acceso a información crítica, preserva la privacidad y hace viable la investigación colaborativa a gran escala, elemento clave para mejorar la predicción, el diagnóstico y el desarrollo de nuevos tratamientos28.
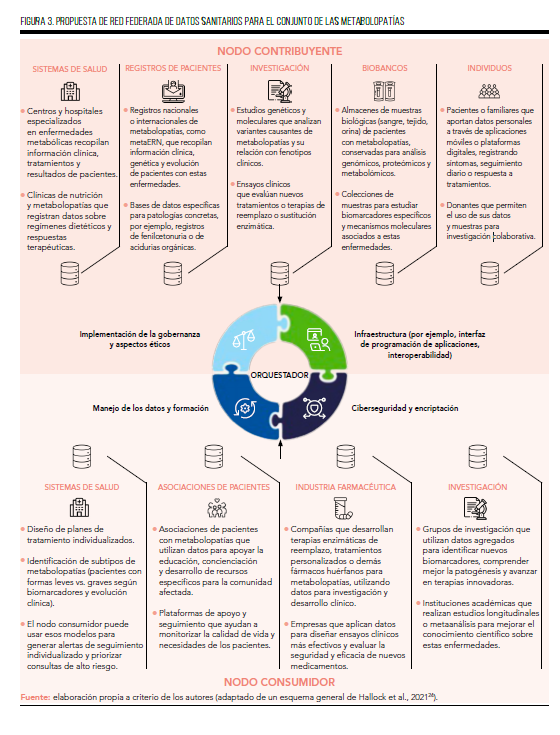
Supongamos ahora un hospital especializado en enfermedades metabólicas como la enfermedad de Fabry (nodo contribuyente) que aporta datos clínicos (grado de dolor neuropático, presencia de angioqueratomas, etc.) y diagnósticos de pacientes; mientras que un registro específico de metabolopatías recopila información longitudinal sobre la evolución de estos pacientes (como el desarrollo o no de insuficiencia cardíaca y/o renal). Los centros de investigación, a su vez, aportan datos genómicos y resultados de estudios experimentales, y los biobancos almacenan muestras biológicas relevantes para análisis posteriores. Incluso los propios pacientes pueden contribuir con datos generados a través de dispositivos de monitorización remota o cuestionarios digitales, aportando información sobre dolor neuropático, fatiga, crisis características, síntomas gastrointestinales o limitaciones funcionales que enriquecen la comprensión real de la enfermedad.
Todos estos datos se coordinan mediante un orquestador que asegura la implementación de normas de gobernanza, proporciona la infraestructura técnica necesaria (interfaz de programación de aplicaciones, interoperabilidad) y garantiza la seguridad y privacidad mediante sistemas de ciberseguridad y encriptación. Gracias a tecnologías como el aprendizaje automático federado, es posible entrenar modelos predictivos que permitan anticipar, por ejemplo, la progresión renal y/o cardíaca utilizando datos distribuidos en múltiples nodos sin necesidad de centralizar la información ni comprometer la privacidad de los pacientes. Esto es especialmente importante en patologías como esta, donde la dispersión y la sensibilidad de los datos dificultan la recopilación masiva en un único repositorio.
Así, los nodos consumidores, que pueden ser otros hospitales, compañías farmacéuticas desarrollando nuevas terapias de sustitución enzimática o equipos de investigación, acceden a modelos de IA mejor entrenados y más robustos, que permiten mejorar la precisión diagnóstica de las metabolopatías, predecir la evolución de la enfermedad o identificar subgrupos de pacientes que responderán mejor a determinadas terapias (como por ejemplo, aquellos con un peor control de la enfermedad con las terapias convencionales). Este enfoque potencia la colaboración y la innovación, acelerando el desarrollo de tratamientos efectivos mientras se mantiene el control y la soberanía sobre los datos.
La Figura 3 recoge una propuesta de red federada de datos sanitarios para el conjunto de las metabolopatías, en la que cada centro puede actuar como nodo contribuyente, aportando datos localmente entrenables, y como nodo consumidor, accediendo a modelos agregados sin exponer información sensible.
Modelos de gobernanza y protección de la privacidad
Los modelos de gobernanza de datos determinan cómo una organización gestiona y controla su información. Al igual que existen distintos tipos de EERR, cada una con sus particularidades, en la gobernanza de datos existen tres modelos principales: centralizado, descentralizado y federado, cada uno con su forma de manejar la información.
En un modelo centralizado, una única autoridad define las políticas, lo que asegura coherencia, pero puede restar agilidad. El modelo descentralizado, en cambio, da autonomía a cada departamento para gestionar sus propios datos, aportando flexibilidad, aunque con riesgo de inconsistencias. El modelo federado combina ambos enfoques: un equipo central marca las reglas generales y los equipos locales las adaptan a sus necesidades, equilibrando coherencia y autonomía.
El creciente uso de la IA en los distintos contextos sanitarios introduce nuevos desafíos de gobernanza, dado que la eficacia de los modelos de IA depende directamente de la calidad de los datos con los que son entrenados. En este sentido, el éxito de los sistemas de IA requiere disponer de datos fiables y de alta calidad, tanto para el entrenamiento de los algoritmos como para la validación de los modelos29.
La Ley de Gobernanza de Datos de la Unión Europea30 impulsa el intercambio de datos mediante marcos y mecanismos de gobernanza bien definidos, fortaleciendo la confianza necesaria para compartir datos sensibles en EERR. Asimismo, el artículo 10 de la Ley de Inteligencia Artificial de la UE31 impone estrictos requisitos en materia de calidad, mitigación de sesgos y transparencia, especialmente relevantes para aplicaciones de IA en el ámbito de las EERR, donde cualquier sesgo o error puede tener un impacto significativo en los pacientes.
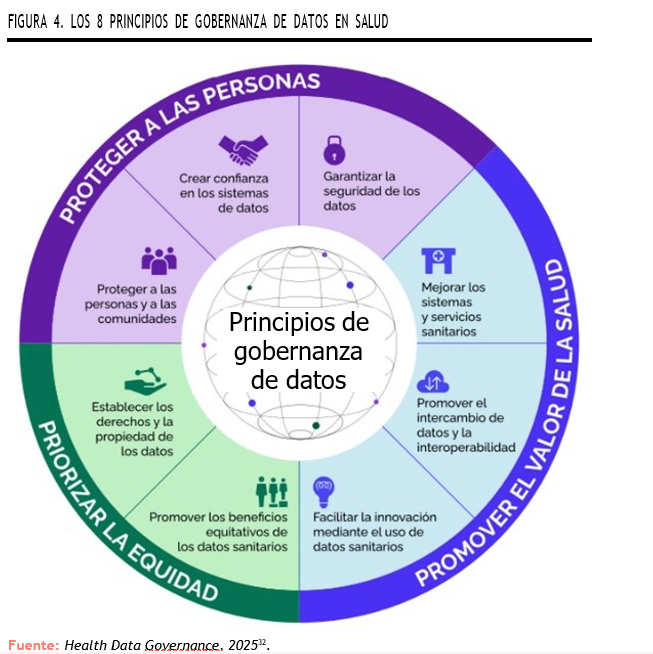
En este contexto, los 8 principios de la Organización para la Cooperación y el Desarrollo Económica (OCDE) sobre gobernanza de los datos de salud ofrecen una base sólida para el desarrollo de marcos nacionales que garanticen una gestión responsable, ética y eficiente de los datos. Algunos de ellos aplican de manera especialmente directa a las EERR debido a sus características particulares, como, por ejemplo, la dispersión geográfica, pues la interoperabilidad permite que los datos de pacientes dispersos se integren y reutilicen, acelerando diagnóstico y tratamiento32.
Iniciativas europeas de espacios de datos sanitarios
A nivel europeo, el Espacio Europeo de Datos Sanitarios (EHDS, por sus siglas en inglés) establece un entorno específico común de datos sanitarios para su uso e intercambio de manera electrónica en toda la UE33. Esta iniciativa busca:
- Empoderar a las personas para que puedan acceder, controlar y compartir sus datos sanitarios electrónicos entre países, con fines asistenciales (uso primario de los datos), mejorando la coordinación en el diagnóstico y tratamiento.
- Permitir la reutilización segura y fiable de los datos de salud con fines de investigación, innovación, formulación de políticas y regulación (uso secundario de los datos).
- Favorecer un mercado único para los sistemas de historia clínica electrónica, que dé soporte tanto al uso primario como al secundario de los datos.
Este espacio está diseñado para beneficiar a todos los ciudadanos de la UE, incluidos los pacientes, los profesionales sanitarios, los investigadores, los responsables políticos y los agentes de la industria (Tabla 2). En el caso de las EERR, el EHDS representa una oportunidad estratégica especialmente relevante, ya que podría facilitar el intercambio transfronterizo de datos clínicos, genómicos y de seguimiento, reduciendo la fragmentación actual y permitiendo que los pacientes se beneficien de diagnósticos más rápidos, tratamientos más coordinados y una investigación más robusta a escala europea.
TABLA 2. PRINCIPALES BENEFICIARIOS DE LA INTRODUCCIÓN DEL EHDS
| Figura |
Ventajas |
| Pacientes |
Acceso rápido y gratuito a sus propios datos, con mayor control, posibilidad de añadir información o restringir el acceso a partes específicas de su historial. Protección de privacidad y seguridad por defecto. |
| Profesionales de la salud |
Acceso más rápido y fácil a los registros de salud en diferentes proveedores de atención médica y países. |
| Investigadores |
Acceso a datos de salud de alta calidad y a gran escala, en un sistema claro y estructurado. |
| Responsables políticos |
Acceso fácil, transparente y rentable a los datos para la mejora de los sistemas sanitarios y la garantía de la seguridad de los pacientes. |
| Industria |
Acceso a datos sanitarios electrónicos anonimizados para investigación aplicada e innovación. |
Fuente: Elaboración propia a partir de European Health Data Space Regulation33
La interoperabilidad como clave para la investigación traslacional
Para alcanzar una gobernanza de datos eficaz en el ámbito de la IA, es esencial que los gobiernos integren y conecten la información procedente de los distintos sistemas sanitarios y sociales. Esto implica crear repositorios de alta calidad, accesibles a investigadores acreditados, junto con entornos analíticos seguros o mecanismos adecuados para la extracción de datos34.
La interoperabilidad es clave para que la información clínica de pacientes con cualquier patología, incluidas las EERR, esté disponible en cualquier centro sanitario, evitando duplicidades en las pruebas, errores en la medicación y pérdidas de información, lo que favorece una atención más coordinada, segura y personalizada. Además, esta conectividad mejora la calidad de los datos usados para investigación y desarrollo de nuevas terapias.
Para ello, se utilizan estándares comunes, como el CEN/ISO 13606 a nivel europeo y herramientas nacionales como el HCDSNS y el REI en España, que armonizan la historia clínica digital y la receta electrónica, respectivamente. La Comisión Europea coordina las infraestructuras transfronterizas, mientras que los distintos organismos reguladores supervisan el cumplimiento normativo, como el Reglamento General de Protección de Datos, y gestionan el acceso y permisos sobre los datos, imponiendo restricciones técnicas según el uso previsto35,36.
Finalmente, el EHDS forma parte de una estrategia de datos más amplia, vinculada a la Ley de Gobernanza de Datos y la Ley de Datos, que establecen obligaciones esenciales para la interoperabilidad y el intercambio seguro de información, especialmente en el ámbito de las EERR 37.
El valor de los datos compartidos en red y plataformas federadas
Biobancos, registros clínicos y fuentes ómicas
Los biobancos, los registros clínicos y las fuentes ómicas (genómica, transcriptómica, proteómica, metabolómica, etc.) permiten contar con cohortes más grandes y, por tanto, un mayor poder estadístico y de descubrimiento en correlaciones entre los datos clínicos y los moleculares. Una revisión sistemática y metaanálisis analizó el impacto de los biobancos en 28 EERR, evidenciando que la integración de la infraestructura de los biobancos y los registros clínicos ayuda a identificar y validar biomarcadores, así como a dilucidar la enfermedad a nivel ómico, favoreciendo así el desarrollo de nuevas estrategias terapéuticas40.
Ejemplo a nivel europeo: RD-Connect39
Un ejemplo paradigmático lo encontramos en la plataforma europea RD-Connect. Esta iniciativa, lanzada en noviembre de 2012, es un recurso global para la investigación de EERR, con el objetivo de superar la fragmentación, inaccesibilidad y aislamiento de los datos característicos de los estudios de las EERR. RD-Connect vincula los datos ómicos con los datos fenotípicos e informaciones de biobancos y registros, permitiendo a los investigadores acceder a ellos más allá de las fronteras institucionales y nacionales, fomentando así investigaciones con una visión más completa e integral.
Ejemplo a nivel nacional: Red Únicas40
La Red Únicas, impulsada en 2023 por el Hospital Sant Joan de Déu (SJD) y la Federación Española de EERR (FEDER), reúne a 30 hospitales para mejorar la atención pediátrica en EERR mediante un modelo colaborativo que incorpora arquitecturas de datos federados. SJD ha consolidado un enfoque asistencial innovador, reforzado por acreditaciones europeas y la creación de la Casa de Sofía, además de iniciativas como ÚNICAS Talks para implicar a las familias.
La Figura 5 representa el modelo integral de atención a las EERR de la Red Únicas, basado en un enfoque centrado en el paciente. Como vemos, uno de los objetivos es utilizar datos e IA para impulsar el diagnóstico de precisión y el desarrollo de nuevas terapias, un enfoque vanguardista que está transformando la atención en este grupo de patologías.
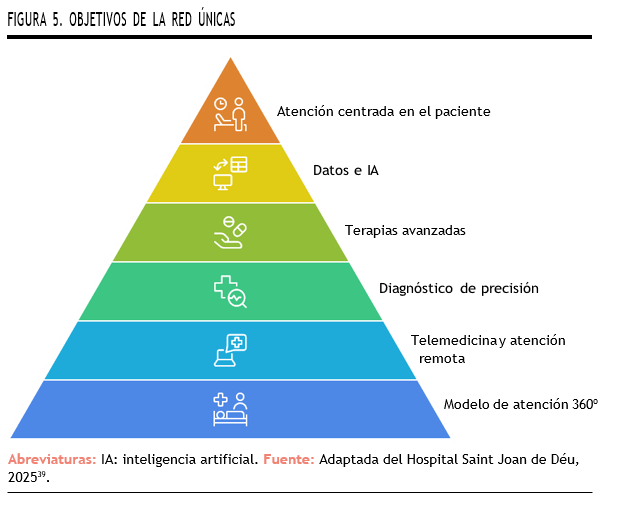
Generación de conocimiento colectivo y colaboración entre centros, pacientes y familias
La participación activa de los pacientes con EERR permite recopilar datos sobre síntomas, calidad de vida y aspectos sociales que rara vez se registran en la práctica clínica. Integrados en plataformas de Big Data, estos datos enriquecen la evidencia, permiten identificar patrones, subtipos de enfermedad y evaluar la efectividad de terapias en condiciones reales. Además, alimentan modelos de IA para predecir progresión y complicaciones, favoreciendo diagnósticos de precisión y tratamientos personalizados, y fortalecen un enfoque centrado en el paciente que guía decisiones clínicas y sanitarias.
La participación activa de los pacientes con EERR permite recopilar datos sobre síntomas, calidad de vida y aspectos sociales que rara vez se registran en la práctica clínica
La Organización Europea para las Enfermedades Raras (EURORDIS) impulsa la Escuela Abierta de Investigación y Desarrollo de Medicamentos de su Academia, una iniciativa destinada a formar tanto a pacientes como a investigadores para que participen de manera informada y activa en la investigación biomédica, especialmente en el ámbito de las enfermedades raras. Al proporcionar a los pacientes conocimientos sobre ensayos clínicos, procesos regulatorios y el uso de Big Data, EURORDIS les permite comprender cómo se recopilan y analizan grandes volúmenes de datos clínicos y PRO (Patient-Reported Outcomes). Esto favorece su implicación en el diseño, seguimiento y evaluación de los estudios, contribuyendo a mejorar la calidad de la investigación y, en última instancia, la atención que reciben las personas afectadas41.
DE LOS DATOS A LA PREDICCIÓN: ANALÍTICA AVANZADA Y MEDICINA PERSONALIZADA
En el contexto de las EERR, la analítica avanzada y el uso de IA permiten extraer patrones significativos y generar modelos predictivos a partir de grandes volúmenes de información clínica y biomédica, superando muchos de los obstáculos metodológicos de la medicina tradicional centrada en estudios restringidos42-44. Sin embargo, la integración de estas herramientas en la clínica y la investigación requiere de una evaluación crítica constante que permita distinguir las correlaciones falsas de los hallazgos robustos y clínicamente relevantes45.
Herramientas de análisis masivo y aprendizaje automático aplicadas a datos clínicos
La analítica masiva en Big Data se apoya en múltiples enfoques estadísticos y computacionales, entre los que destacan el aprendizaje automático (ML, machine learning) y el aprendizaje profundo (DL, deep learning). Los modelos de ML permiten identificar relaciones complejas entre variables, segmentar pacientes en subgrupos homogéneos y predecir desenlaces a partir de datos heterogéneos (incluyendo clínicos, genéticos y sociales)45,46.
En el entorno de las EERR, estas herramientas ayudan a mejorar la detección de fenotipos poco reconocidos, optimizar el reclutamiento para ensayos clínicos y personalizar terapias, aunque las exigencias de calidad y representatividad de datos permanecen como un reto principal44,47. Recientes avances demuestran que los algoritmos de ML y DL, como redes neuronales y random forests, pueden alcanzar altas tasas de sensibilidad y especificidad en tareas como la predicción de reacciones adversas medicamentosas, la estratificación de pacientes según riesgo y el ajuste de la dosis farmacológica46,47. Particularmente relevante ha sido la aplicación de modelos conversacionales, basados en grandes modelos de lenguaje (LLM, large language models), que permiten simular diálogos clínicos, facilitar el triaje y mejorar la toma de decisiones diagnósticas en escenarios complejos. No obstante, estas técnicas presentan riesgos inherentes: el sobreajuste a datos de entrenamiento, la falta de interpretabilidad y la generalización limitada cuando se aplican a poblaciones que difieren de los conjuntos originales. Se requiere la intervención continua de expertos clínicos para validar las variables empleadas, la revisión crítica de los resultados y la atención a posibles sesgos representativos44,49,50.
Modelos predictivos de progresión y respuesta terapéutica
Otra aportación del Big Data a las EERR es el desarrollo de modelos predictivos que anticipan la evolución clínica y la respuesta a intervenciones terapéuticas. Mediante el procesamiento de datos longitudinales, electrónicos, genómicos o de registros de pacientes, es posible estimar trayectorias individuales de progresión y evaluar la utilidad directa de diferentes tratamientos51.
Ejemplos recientes en EERR incluyen el uso de ML para estimar el riesgo de complicaciones hematológicas en enfermedades como el síndrome de Shwachman-Diamond a partir de biomarcadores y datos genéticos, y el diseño de modelos de respuesta farmacológica personalizados en base a perfiles genómicos y registros históricos52. Estos enfoques permiten la optimización de recursos asistenciales y, potencialmente, una mejora en la calidad de vida de los pacientes.
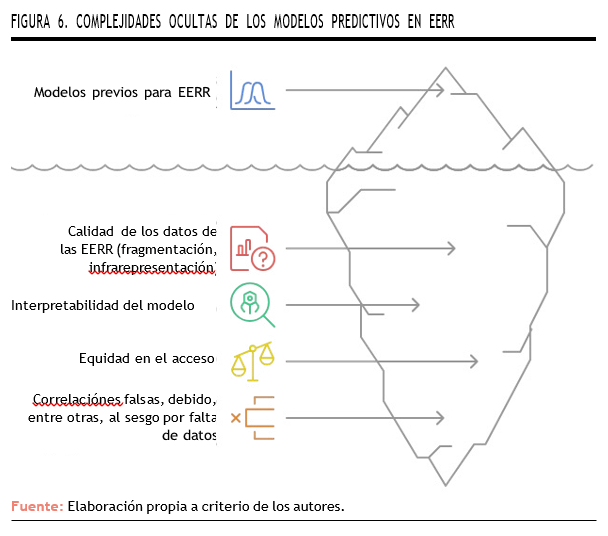
Sin embargo, la predicción siempre debe ser contextualizada. La variabilidad genética y fenotípica de las EERR implica que la validez externa y la reproducibilidad pueden quedar comprometidas, y parte de los datos disponibles provienen de muestras reducidas o altamente seleccionadas. Dicha limitación epistemológica obliga a incorporar métodos bayesianos y diseño adaptativo que permitan la reevaluación continua del modelo predictivo conforme se acumula nueva evidencia53,54 (Figura 6).
Aplicaciones en la prevención, diagnóstico temprano y estratificación de pacientes
El impacto más inmediato de la analítica avanzada en el campo de las EERR se observa en tres ámbitos: prevención, diagnóstico temprano y estratificación de pacientes. Los sistemas de IA pueden analizar información rutinaria (historias clínicas electrónicas, datos de laboratorio, registros de actividad) para identificar patrones sugestivos de enfermedad antes de la aparición de síntomas clínicos evidentes, acelerando el acceso al diagnóstico y permitiendo una intervención precoz55.
En la estratificación de pacientes, el clustering mediante aprendizaje no supervisado ha servido para distinguir subgrupos de pacientes que responden de forma diferente a tratamientos estándar, así como para establecer perfiles pronósticos en enfermedades de curso incierto56-58. Estas estrategias han sido cruciales en la medicina personalizada y en la organización de cohortes para ensayos clínicos adaptativos.
A pesar de sus beneficios, es necesario considerar que la adopción de estas herramientas puede amplificar desigualdades si no se diseñan con criterios de equidad y respeto a la diversidad poblacional. La calidad de los datos, la transparencia en los modelos y el acceso igualitario a estas tecnologías deben ser prioritarios para evitar sesgos y riesgos éticos, tal como han señalado autores recientes y organismos internacionales58,59.
HERRAMIENTA ANALÍTICA FARMACIA12
Desarrollada por el Ministerio de Sanidad de España con el objetivo de apoyar la toma de decisiones sobre reembolso, precio y cobertura de medicamentos en el sistema público, su finalidad principal es reducir la incertidumbre clínica y económica asociada a la incorporación de nuevas terapias, anticipando su impacto presupuestario y facilitando la gestión estratégica de los recursos sanitarios.
Analítica Farmacia integra múltiples fuentes de información, incluyendo datos internos de consumo y facturación, modelos de precio y gasto, literatura científica publicada en ensayos clínicos y congresos, así como “literatura gris” como informes o notas de prensa.
A partir de toda esta información, la herramienta utiliza técnicas de inteligencia artificial y análisis avanzado de datos para generar predicciones y escenarios futuros sobre la financiación y utilización de medicamentos, permitiendo evaluar diferentes estrategias y riesgos asociados.
Entre sus aplicaciones previstas destacan la evaluación de nuevas tecnologías y fármacos para su posible financiación pública, la modelización de costes y beneficios, la estimación de techos de gasto o esquemas de riesgos compartidos, y el apoyo a la política farmacéutica, contribuyendo a la sostenibilidad del sistema y a un acceso más equitativo a los tratamientos.
Su desarrollo surge en el contexto de la pandemia de COVID-19, tras comprobar la utilidad de grandes volúmenes de datos para la planificación sanitaria, y busca aplicar de manera sistemática la inteligencia artificial en la gestión de la prestación farmacéutica pública.
En términos de impacto, Analítica Farmacia podría mejorar la eficiencia del sistema sanitario, agilizar la toma de decisiones sobre reembolso, aumentar la predictibilidad en la incorporación de fármacos y favorecer un uso más racional y sostenible de los recursos públicos, contribuyendo así a una mejor disponibilidad de terapias para los pacientes.
Implicaciones éticas y regulatorias del uso de IA en salud
Con el avance del conocimiento y de la tecnología surgen nuevos desafíos éticos y legales, especialmente cuando la legislación y la integración social no evolucionan al mismo ritmo que la innovación. Históricamente, los datos de salud se encontraban aislados y fragmentados, con un consentimiento insuficiente para su uso y con preocupaciones sobre la privacidad y el anonimato de los pacientes60. La creación de bases de datos de salud centralizadas ayudó a sortear algunos de estos problemas, como la anonimización de los datos y mejora de la interoperabilidad, como en The European Genome-phenome Archive61. Sin embargo, la heterogeneidad de los datos, su protección y la falta de infraestructuras adecuadas continuaron siendo desafíos críticos para una sanidad basada en Big Data.
En respuesta, las bases de datos descentralizadas buscan reducir riesgos para la privacidad y la autonomía personal, al tiempo que refuerzan la transparencia, la confianza y la equidad62. En este contexto, Skovgaard et al.63 mostraron que los ciudadanos de la UE apoyan la reutilización de datos sanitarios para fines distintos al tratamiento, siempre que contribuyan al bien común. El Reglamento General de Protección de Datos (RGPD) intenta equilibrar la privacidad con la necesidad de compartir datos con fines sanitarios y de investigación. En la misma línea regulatoria, la Ley de Gobernanza de Datos de la UE promueve el intercambio de datos mediante marcos claros de gobernanza que refuercen la confianza y la transparencia29, mientras que el artículo 10 de la Ley de Inteligencia Artificial de la UE establece estrictos requisitos de calidad, mitigación de sesgos y transparencia para aplicaciones de IA de alto riesgo64. No obstante, persisten preocupaciones sobre la comercialización, la seguridad y el uso potencialmente perjudicial de los datos especialmente en EERR, ya que, dada la escasez de datos de estos pacientes y la alta sensibilidad de su información genética y clínica, los sesgos algorítmicos provocan sesgos hacia la mayoría de los datos, pudiendo dar resultados injustos o no representativos. En este escenario, la ética y la regulación deben adaptarse para permitir el aprovechamiento de Big Data e IA en beneficio de los pacientes, garantizando al mismo tiempo la protección y la equidad.
BIG DATA Y EVALUACIÓN ECONÓMICA DE INTERVENCIONES EN EERR
La incorporación del Big Data y los datos del mundo real (Real-World Data, RWD) en la evaluación económica de intervenciones dirigidas a EERR está transformando de manera significativa el modo en que se generan, analizan y utilizan las evidencias para la toma de decisiones sanitarias65.
Las características propias de estas patologías (principalmente, la baja prevalencia, la heterogeneidad clínica y la escasez de datos) han generado históricamente limitaciones metodológicas importantes en herramientas de evaluación económica, como los análisis coste-efectividad y coste-utilidad, o en análisis de impacto presupuestario. La progresiva aparición de nuevas fuentes de datos, infraestructuras de información interoperables y herramientas analíticas avanzadas abre una oportunidad sin precedentes para mejorar la calidad de la evidencia, reducir la incertidumbre y favorecer decisiones más equitativas y eficientes.
La Tabla 3 presenta ejemplos potenciales del uso de Big Data en el contexto de la evaluación de tecnologías sanitarias para EERR, mostrando cómo los datos masivos pueden apoyar el análisis de coste-efectividad, la generación de evidencia del mundo real, la estimación del impacto presupuestario, la valoración del valor social de las intervenciones y la facilitación de procesos y acuerdos de negociación.
TABLA 3. POTENCIAL DEL BIG DATA EN LA EVALUACIÓN DE TECNOLOGÍAS SANITARIAS EN EL CONTEXTO DE LAS EERR
| Línea Estratégica |
Descripción |
Ejemplos en EERR |
| Fortalecer análisis de coste-efectividad y coste-utilidad |
Uso de datos reales y técnicas avanzadas para mejorar parámetros clave de los modelos económicos. |
Posibilita estimar parámetros fundamentales para los modelos económicos, como:
• Incidencia y prevalencia: uso de registros nacionales de distrofias musculares para estimar la incidencia real de Duchenne.
• Patrones de progresión: modelos predictivos que anticipan la pérdida de la marcha en AME.
• Uso de recursos sanitarios: análisis de historias clínicas electrónicas para medir hospitalizaciones y uso de ventilación no invasiva en enfermedades neuromusculares.
• Desenlaces clínicos: IA para identificar biomarcadores tempranos de progresión en la enfermedad de Fabry. |
| Evaluar el impacto presupuestario basado en datos reales |
Uso de algoritmos y bases de datos clínicos para identificar población elegible y proyectar el gasto real. |
• Identificación automatizada de pacientes con mutaciones específicas en fibrosis quística para terapias moduladoras.
• Uso de registros de errores congénitos del metabolismo para estimar cuántos pacientes serían candidatos a nuevas terapias génicas.
• Monitorización del gasto real en MMHH utilizando paneles de control integrados en tiempo real. |
| Añadir valor social y equidad en la toma de decisiones |
Incorporación de dimensiones sociales y análisis de desigualdades en el acceso. |
• Medición de la carga del cuidador en enfermedades como epidermólisis bullosa (tiempo de curas diarias, impacto laboral).
• Identificación de desigualdades regionales en el acceso al diagnóstico genético precoz de AME.
• Análisis de diferencias socioeconómicas en la supervivencia o calidad de vida en HTP. |
| Facilitar los procesos de negociación |
Monitorización de resultados en vida real para acuerdos de pago por resultados. |
• Acuerdos de pago por efectividad real en terapias avanzadas para AME mediante seguimiento de resultados motores (sentarse, caminar).
• Evaluación en tiempo real de biomarcadores renales en Enfermedad de Fabry para ajustar pagos según respuesta.
• Modelos de riesgo compartido para terapias génicas en hemoglobinopatías raras basados en necesidad de transfusiones a largo plazo. |
Abreviaturas: AME: atrofia muscular espinal; HTP: hipertensión pulmonar; IA: inteligencia
artificial; MMHH: medicamentos huérfanos. Fuente: elaboración propia a criterio de los
autores.
CASOS DE USO Y EXPERIENCIAS INTERNACIONALES
En los últimos años han surgido numerosas iniciativas internacionales, lideradas por la UE, Redes Europeas de Referencia, grandes consorcios de investigación y proyectos específicos por patología, que permiten analizar datos distribuidos sin necesidad de centralizarlos, garantizando interoperabilidad, privacidad y cumplimiento ético-legal. Estas iniciativas combinan enfoques como aprendizaje federado, consultas semánticas distribuidas, armonización FAIR y federación de registros, y han demostrado mejoras significativas en diagnóstico, descubrimiento biomédico y estandarización de recursos (Tabla 4).
TABLA 4. EJEMPLOS DE INICIATIVAS INTERNACIONALES BASADAS EN DATOS FEDERADOS EN EL ÁMBITO DE LAS EERR
| Iniciativa/Proyecto |
Ámbito y Objetivo |
Modelo de Datos |
Participantes Clave |
Resultados Relevantes |
Presupuesto |
| GENERALES |
| Solve-RD66 |
Diagnóstico de EERR mediante datos de múltiples países y análisis “-ómica”. |
Infraestructura de datos compartidos pan-europea con acceso controlado; estandarización y reanálisis masivo de datos clínicos y genómicos. Aunque no siempre literalmente “federada”, promueve la interoperabilidad entre recursos distribuidos. |
Más de 21.000 conjuntos de datos de familias afectadas por EERR, múltiples ERN implicadas. |
Mejora del diagnóstico (casos resueltos): > 5000 familias analizadas. La agregación de datos y la interoperabilidad estructural son fundamentales para avanzar en EERR; plantea retos de gobernanza, consentimiento, calidad de datos. |
Total: 15.361.621 € |
| European Joint Programme on Rare Diseases (EJP-RD)67 |
Integrar recursos de investigación y datos de EERR a través de una plataforma federada. |
Plataforma VP-RD que conecta repositorios nacionales y de ERN; interoperabilidad FAIR. |
>130 instituciones; 24 ERN; EURORDIS. |
Ha demostrado la viabilidad del acceso federado en investigación de EERR. Importancia del consentimiento dinámico. |
Total: 100.655.230 € (Contribución UE: 55.073.831 €) |
| ELIXIR Rare Diseases68 |
Crear una infraestructura federada que permita a los investigadores descubrir, acceder y analizar distintos repositorios de EERR en toda Europa. |
Enfoque federado de los datos: integración de repositorios, registros, biobancos y datos -ómicos; aplicación de los principios FAIR a los datos de EERR. |
11 países europeos y alianzas con otros actores europeos en EERR como EURORDIS, RD-Connect; bbmri-eric. |
Catálogo actualizado de herramientas y recursos para investigación en enfermedades raras (mantenido en bio.tools). Desarrollo de métricas FAIR específicas. Pilotaje de infraestructura federada. |
– |
| ESPECÍFICOS PARA UNA ENFERMEDAD O GRUPO DE ENFERMEDADES |
| SYNTHEMA69 |
Crear un hub de datos transfronterizo para enfermedades hematológicas raras, con foco en anemia drepanocítica y leucemia mieloide aguda. |
Infraestructura de aprendizaje federado + computación multipartita segura + privacidad diferencial para entrenamiento y generación de datos sintéticos sin mover los datos crudos. |
16 socios en 10 países europeos; coordinado por la UPM (España). Colaboración con ERN-EuroBloodNet. |
Supera el problema de escasez y fragmentación de datos en hematologías raras mediante generación de “pacientes virtuales”. Arquitectura técnica de federación + anonimizaciones. |
6.514.560 € |
| Euro-NMD Registry hub70 |
Recopilar datos de todos los pacientes neuromusculares atendidos por los 82 centros sanitarios pertenecientes a la ERN Euro-NMD. |
Plataforma de registro hub federada: se conecta con múltiples registros específicos mediante interoperabilidad, siguiendo el principio de “data visiting” y principios FAIR. |
82 centros sanitarios de 25 países europeos (ERN Euro-NMD) y 27 organizaciones de pacientes. |
Ejemplo de infraestructura federada que permite análisis sin transferencia de datos. Empoderamiento del paciente, interoperabilidad semántica. |
Financiación UE: 200.000 €. Contribución especie pacientes: 274.641 €. |
| FAIRVASC71 |
Crear un acceso FAIR a una red de registros de vasculitis asociada a ANCA. |
Tecnologías semánticas y consultas federadas: datos locales convertidos a RDF y enlazados mediante ontología común. Consultas SPARQL sin mover datos crudos. |
Registros nacionales/regionales de vasculitis de 7 países (Irlanda, Reino Unido, Francia, Alemania, Suecia, República Checa y Polonia). |
Análisis federado de >3.800 pacientes. Identificación de 5 subgrupos clínicos y serológicos de la enfermedad. Ha demostrado la viabilidad de consultas federadas en EERR. |
2.299.091 € |
Abreviaturas: ANCA: anticuerpos anticitoplasma de neutrófilos; EERR: enfermedades raras; ERN: redes europeas de referencia (European
Reference Networks). FAIR: Encontrables, Accesibles, Interoperables y Reutilizables (Findable, Accessible, Interoperable, Reusable); RDF: Resource
Description Framework; UE: Unión Europea. Fuente: referencias bibliográficas incluidas en la tabla.En España, el uso de modelos de datos federados y arquitecturas distribuidas está cobrando un papel cada vez más relevante para abordar los retos de las EERR.
En España, el uso de modelos de datos federados y arquitecturas distribuidas está cobrando un papel cada vez más relevante para abordar los retos de las EERR
Aunque el país no dispone todavía de una infraestructura nacional plenamente federada, sí existen varias iniciativas consolidadas, como CIBERER, BioNER, la Red de Enfermedades Raras del Centro Superior de Investigaciones Científicas (CSIC) o los registros autonómicos y nacionales coordinados por el ISCIII, que ya aplican estrategias de interoperabilidad avanzada, mantenimiento distribuido de datos, acceso controlado o armonización FAIR (Encontrables, Accesibles, Interoperables y Reutilizables, por sus siglas en inglés).
Además, España lidera proyectos europeos de referencia en aprendizaje federado en salud, como SYNTHEMA69, situándose en una posición destacada en la creación de ecosistemas de datos seguros, integrados y colaborativos. En conjunto, estas iniciativas demuestran un progreso sostenido hacia un modelo federado real que facilite el diagnóstico, la investigación traslacional y la planificación sanitaria en EERR.
OPORTUNIDADES FUTURAS Y CONCLUSIONES
El uso de Big Data en el ámbito de las EERR abre un escenario de enormes oportunidades, pero también plantea desafíos significativos que deben abordarse de manera coordinada. La creación de espacios de datos sanitarios interoperables y seguros exige superar obstáculos técnicos, como la heterogeneidad de fuentes, la falta de estándares comunes o la escasez de datos de alta calidad, junto con barreras legales y éticas relacionadas con la privacidad, el consentimiento dinámico y la gobernanza del uso secundario de la información.
La integración segura de los datos clínicos, genómicos, de biobancos y de vida real permitiría generar cohortes virtuales más amplias y representativas, acelerando el diagnóstico, el desarrollo de modelos predictivos y la identificación de subgrupos de pacientes que respondan mejor a determinadas terapias.
A la vez, estos espacios pueden convertirse en infraestructuras sostenibles si incorporan mecanismos robustos de calidad, modelos federados, semántica común, auditoría en tiempo real y una financiación continuada. El potencial de generar valor económico y clínico a partir de datos actualizados y con flujos continuos permite, además, avanzar hacia evaluaciones de impacto más precisas, decisiones regulatorias ágiles y sistemas de salud más eficientes, que facilitarían estimaciones más robustas de carga de la enfermedad, costes y efectividad comparada, reduciendo la incertidumbre inherente a la toma de decisiones sobre MMHH. Además, permitirían monitorizar resultados en práctica real y apoyar modelos de pago por resultados.
En conjunto, estas transformaciones apuntan a un futuro en el que las personas con EERR se beneficien de una medicina verdaderamente personalizada, preventiva y participativa, sustentada en datos confiables y accesibles. Para ello se necesita un impulso decidido de políticas públicas que fomenten la interoperabilidad, la transparencia y la equidad, así como una colaboración abierta entre administraciones, profesionales, industria, comunidad investigadora y asociaciones de pacientes. Solo mediante esta convergencia será posible democratizar el acceso al conocimiento, acelerar el diagnóstico y mejorar la calidad de vida de quienes viven con este tipo de patologías.
REFERENCIAS
- Pastorino R, De Vito C, Migliara G, et al. Benefits and challenges of Big Data in healthcare: an overview of the European initiatives. Eur J Public Health. 2019;29(Supplement_3):23-27.
- Manyika J, Chui M, Brown B, et al. Big data: The next frontier for innovation, competition, and productivity. 2011. Disponible en: www.mckinsey.com/mgi
- Beyer M, Laney D. The Importance of ‘Big Data’: A Definition. Gartner report. 2012.
- McAfee A, Brynjolfsson E. Big data: the management revolution. Harv Bus Rev. 2012;90(10):60-66, 68, 128.
- Yao L, Liu Y, Wang T, et al. Global trends of big data analytics in health research: a bibliometric study. Front Med. 2025;12:1456286.
- World Economic Forum. How to harness the power of health data to improve patient outcomes. 2024.
- Alsahfi T, Badshah A, Aboulola OI, Daud A. Optimizing healthcare big data performance through regional computing. Sci Rep. 2025;15(1):3129.
- Badshah A, Daud A, Alharbey R, Banjar A, Bukhari A, Alshemaimri B. Big data applications: overview, challenges and future. Artif Intell Rev. 2024;57(11):290.
- World Health Organization, World Health Organization – Europe. Impact training for Big Data in health care. 2024.
- European Comission. €204 million funding for projects boosting innovation in business digitalisation, digital skills, health, public and automotive sectors. 2025.
- Agència de Qualitat i Avaluació Sanitàries de Catalunya (AQuAS). Guía de evaluación de tecnologías de salud digital que incorporan inteligencia artificial (IA). 2024.
- Arganda C. Sanidad se sube al carro de ChatGPT y las IA para gestionar el precio y reembolso. Diariofarma. 2024.
- Raghupathi W, Raghupathi V. Big data analytics in healthcare: promise and potential. Health Inf Sci Syst. 2014;2(1):3.
- Taruscio D, Floridia G, Salvatore M, Groft SC, Gahl WA. Undiagnosed Diseases: Italy-US Collaboration and International Efforts to Tackle Rare and Common Diseases Lacking a Diagnosis. In: Posada De La Paz M, Taruscio D, Groft SC, eds. Rare Diseases Epidemiology: Update and Overview. Vol 1031. Advances in Experimental Medicine and Biology. Springer International Publishing. 2017:25-38.
- Fermaglich LJ, Miller KL. A comprehensive study of the rare diseases and conditions targeted by orphan drug designations and approvals over the forty years of the Orphan Drug Act. Orphanet J Rare Dis. 2023;18(1):163.
- Degtiar I. A review of international coverage and pricing strategies for personalized medicine and orphan drugs. Health Policy. 2017;121(12):1240-1248.
- Julkowska D, Austin CP, Cutillo CM, et al. The importance of international collaboration for rare diseases research: a European perspective. Gene Ther. 2017;24(9):562-571.
- Heath G, Farre A, Shaw K. Parenting a child with chronic illness as they transition into adulthood: A systematic review and thematic synthesis of parents’ experiences. Patient Educ Couns. 2017;100(1):76-92.
- Adams LS, Miller JL, Grady PA. The Spectrum of Caregiving in Palliative Care for Serious, Advanced, Rare Diseases: Key Issues and Research Directions. J Palliat Med. 2016;19(7):698-705.
- Tumiene B, Graessner H. Rare disease care pathways in the EU: from odysseys and labyrinths towards highways. J Community Genet. 2021;12(2):231-239.
- Agencia Española de Protección de Datos. APROXIMACIÓN A LOS ESPACIOS DE DATOS DESDE LA PERSPECTIVA DEL RGPD. 2023.
- Ministerio para la Transformación Digital y de la Fundación Pública. ¿Por qué espacios de datos? 2024.
- Ministerio para la Transformación Digital y de la Fundación Pública. Aprendizaje automático federado: generación de valor a partir de datos compartidos manteniendo la privacidad. 2025.
- Hallock H, Marshall SE, ’T Hoen PAC, et al. Federated Networks for Distributed Analysis of Health Data. Front Public Health. 2021;9:712569.
- Xu J, Glicksberg BS, Su C, Walker P, Bian J, Wang F. Federated Learning for Healthcare Informatics. J Healthc Inform Res. 2021;5(1):1-19.
- Pati S, Baid U, Zenk M, et al. The Federated Tumor Segmentation (FeTS) Challenge. arXiv. Preprint online 2021.
- AI4VBH – AI Centre for Value Based Healthcare. AI technology provides the opportunity to target diverse clinical pathways, from head to toe, in early life and old age. 2025.
- Ministerio para la Transformación Digital y de la Fundación Pública. Aprendizaje automático federado: generación de valor a partir de datos compartidos manteniendo la privacidad. datos.gob.es. 2025.
- Ethics and Governance of Artificial Intelligence for Health: WHO Guidance. 1st ed. World Health Organization; 2021.
- Off J Eur Union. Regulation (EU) 2022/868 of the European Parliament and of the Council of 30 May 2022 on European Data Governance and Amending Regulation (EU) 2018/1724. 2022.
- Off J Eur Union. Regulation (EU) 2024/1689 of the European Parliament and of the Council of 13 June 2024 Laying down Harmonised Rules on Artificial Intelligence. 2024.
- OECD. Health Data Governance for the Digital Age: Implementing the OECD Recommendation on Health Data Governance. OECD Publishing. 2022.
- European Comission. European Health Data Space Regulation (EHDS). 2025.
- Morley J, Murphy L, Mishra A, Joshi I, Karpathakis K. Governing Data and Artificial Intelligence for Health Care: Developing an International Understanding. JMIR Form Res. 2022;6(1):e31623.
- Carrero Muñoz A. Las normas europeas de la HCE CEN/ISO 13606. 2025.
- Ministerio de Sanidad. Historia Clínica del Sistema Nacional de Salud (HCDSNS). 2025.
- Comisión Europea. Reglamento relativo al Espacio Europeo de Datos de Salud (EEDS). 2025.
- European Commission. RD-CONNECT: An integrated platform connecting registries, biobanks and clinical bioinformatics for rare disease research. CORDIS. 2024.
- Sant Joan de Dèu Barcelona Hospital. Únicas, atención a las enfermedades raras. 2025.
- Garcia M, Downs J, Russell A, Wang W. Impact of biobanks on research outcomes in rare diseases: a systematic review. Orphanet J Rare Dis. 2018;13(1):202.
- EURODIS. EURODIS Open Academy. 2025.
- He D, Wang R, Xu Z, et al. The use of artificial intelligence in the treatment of rare diseases: A scoping review. Intractable Rare Dis Res. 2024;13(1):12-22.
- Rennie O. Navigating the uncommon: challenges in applying evidence-based medicine to rare diseases and the prospects of artificial intelligence solutions. Med Health Care Philos. 2024;27(3):269-284.
- Siderius L, Perera SD, Jankauskaite L, Bhattacharya A, Gonçalves P. Rare diseases: ethical challenges in the era of digital health. Front Digit Health. 2025;7:1539841.
- Bestard Cassinello A. Debilidades y fortalezas del uso de inteligencia artificial para el diagnóstico precoz de enfermedades raras. Una revisión bibliográfica. 2025.
- Tu T, Schaekermann M, Palepu A, et al. Towards conversational diagnostic artificial intelligence. Nature. 2025;642(8067):442-450.
- Abbas SR, Abbas Z, Zahir A, Lee SW. Advancing genome-based precision medicine: a review on machine learning applications for rare genetic disorders. Brief Bioinform. 2025;26(4):bbaf329.
- Germain DP, Gruson D, Malcles M, Garcelon N. Applying artificial intelligence to rare diseases: a literature review highlighting lessons from Fabry disease. Orphanet J Rare Dis. 2025;20(1):186.
- Wojtara M, Rana E, Rahman T, Khanna P, Singh H. Artificial intelligence in rare disease diagnosis and treatment. Clin Transl Sci. 2023;16(11):2106-2111.
- Beam AL, Manrai AK, Ghassemi M. Challenges to the Reproducibility of Machine Learning Models in Health Care. JAMA. 2020;323(4):305.
- James KN, Phadke S, Wong TC, Chowdhury S. Artificial Intelligence in the Genetic Diagnosis of Rare Disease. Clin Lab Med. 2023;43(1):127-143.
- Trognon A. Computational diagnosis of Shwachman-Diamond syndrome through cognitive and dialogical investigations. 2022.
- Banerjee J, Taroni JN, Allaway RJ, Prasad DV, Guinney J, Greene C. Machine learning in rare disease. Nat Methods. 2023;20(6):803-814.
- Alzahrani AA, Alharithi FS. Machine learning approaches for advanced detection of rare genetic disorders in whole-genome sequencing. Alex Eng J. 2024;106:582-593.
- Bogart KR, Dermody SS. Relationship of rare disorder latent clusters to anxiety and depression symptoms. Health Psychol. 2020;39(4):307-315.
- Chen X, Wang J, Faviez C, et al. An Integrated Pipeline for Phenotypic Characterization, Clustering and Visualization of Patient Cohorts in a Rare Disease-Oriented Clinical Data Warehouse. In: Mantas J, Hasman A, Demiris G, et al., eds. Studies in Health Technology and Informatics. IOS Press. 2024.
- Zhu Q, Nguyen DT, Sheils T, et al. Scientific evidence based rare disease research discovery with research funding data in knowledge graph. Orphanet J Rare Dis. 2021;16(1):483.
- Jacoba CMP, Celi LA, Lorch AC, et al. Bias and Non-Diversity of Big Data in Artificial Intelligence: Focus on Retinal Diseases. Semin Ophthalmol. 2023;38(5):433-441.
- Lee J, Liu C, Kim J, et al. Deep learning for rare disease: A scoping review. J Biomed Inform. 2022;135:104227.
- Powell K. The broken promise that undermines human genome research. Nature. 2021;590(7845):198-201.
- The European Genome-phenome Archive (EGA). 2025.
- Ienca M, Ferretti A, Hurst S, Puhan M, Lovis C, Vayena E. Considerations for ethics review of big data health research: A scoping review. PLOS ONE. 2018;13(10):e0204937.
- Skovgaard LL, Wadmann S, Hoeyer K. A review of attitudes towards the reuse of health data among people in the European Union: The primacy of purpose and the common good. Health Policy. 2019;123(6):564-571.
- European Comission. Artículo 10: Datos y Gobernanza de Datos. 2026.
- Liu J, Barrett JS, Leonardi ET, et al. Natural History and Real-World Data in Rare Diseases: Applications, Limitations, and Future Perspectives. J Clin Pharmacol. 2022;62 Suppl 2(Suppl 2):S38-S55.
- Zurek B, Ellwanger K, Vissers LELM, et al. Solve-RD: systematic pan-European data sharing and collaborative analysis to solve rare diseases. Eur J Hum Genet. 2021;29(9):1325-1331.
- EURODIS RARE DISEASE EUROPE. European Joint Programme on Rare Diseases. EJP on RD.
- ELIXIR. Rare Diseases Community.
- SYNTHEMA. SYNTHEMA.
- EURO-NMD. Registry Hub for Rare Neuromuscular Diseases.
- FAIRVASC.










")




