Fernando Abdalla, Irene Fernández y Carlos Dévora
Departamentos de Health Affairs & Policy Research y Health Economics & Market Access, Vivactis Weber
Las enfermedades raras (EERR) suponen un desafío significativo para la salud pública, afectando a entre el 4 % y el 8 % de la población mundial1. A pesar de los avances en conocimiento y tecnología, la diversidad en la manifestación y causa de estas más de 5.000 patologías dificulta en muchos casos su diagnóstico temprano. Los retrasos diagnósticos causan sufrimiento a los pacientes y sus familias, además de imponer un alto coste al sistema de salud y aumentar el riesgo de complicaciones. En España, el 57 % de las personas que padecen EERR experimenta una demora considerable en su diagnóstico, con un tiempo medio superior a los 6 años2.
En este contexto, el cribado neonatal y la detección temprana juegan un papel fundamental en el impacto de las EERR, dado que aproximadamente el 72 % de ellas tienen un origen genético y alrededor del 70 % se manifiestan durante la infancia1. Implementar programas que identifiquen dichos trastornos en etapas iniciales es crucial, ya que permiten intervenciones y tratamientos oportunos, mejorando así la calidad de vida de las personas afectadas. Además, la detección temprana impulsa el desarrollo de terapias específicas y avances en investigación clínica, brindando esperanza a quienes se enfrentan a este tipo de patologías. En última instancia, el cribado neonatal y la detección precoz no solo mejoran el manejo de las EERR, sino que también contribuyen a un futuro más prometedor para los pacientes.
Este artículo está estructurado en tres secciones fundamentales. En primer lugar, abordaremos el proceso diagnóstico en EERR, haciendo hincapié en las diferencias entre el cribado neonatal y el diagnóstico temprano, así como los beneficios asociados con la detección precoz de estas condiciones. La segunda sección examinará los desafíos que enfrentamos, los avances científicos realizados hasta la fecha y las posibles soluciones propuestas para mejorar la detección temprana de las EERR. En la última parte, nos enfocaremos en el análisis del cribado neonatal, evaluando la situación actual, el acceso y las barreras existentes en España, y presentando las mejores prácticas y recomendaciones políticas identificadas hasta el momento. En conjunto, ofreceremos una visión de la importancia y los aspectos clave de la detección temprana de las EERR.
Las enfermedades raras son un desafío para la salud pública debido a su diversidad y retrasos diagnósticos
La relevancia del diagnóstico temprano en EERR
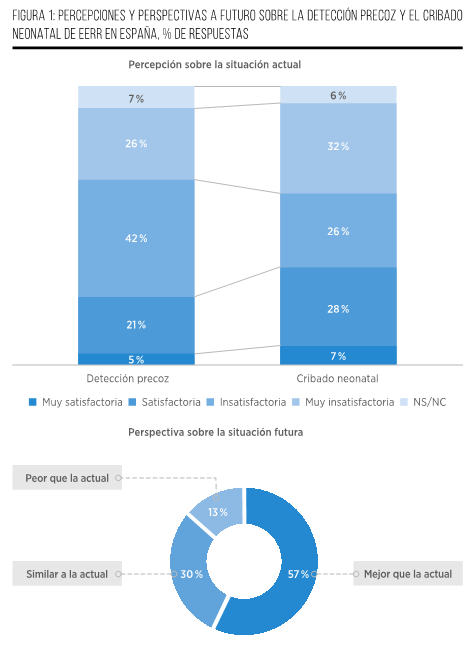
El diagnóstico temprano y el cribado neonatal de EERR son conceptos que, aunque distintos, están interrelacionados y desempeñan un papel crucial en el manejo de enfermedades poco comunes (Figura 1).
El diagnóstico precoz se enfoca en detectar tempranamente la presencia de una EERR en un individuo que ya presenta síntomas o signos indicativos de la misma. Esta identificación de la enfermedad puede tener lugar en cualquier etapa de la vida (no se restringe por tanto al periodo neonatal), y generalmente requiere de la realización de pruebas específicas, como análisis de sangre, pruebas genéticas o resonancias magnéticas, para confirmar el diagnóstico en el paciente3.
En general, el diagnóstico temprano es un proceso más largo que el cribado neonatal y comienza en el momento en que aparecen los síntomas, lo cual suele llevar a una primera consulta médica en un centro de atención primaria. Posteriormente, se desarrolla el periodo que abarca desde la primera consulta médica hasta la derivación a una atención especializada. Por último, el periodo final comprende desde la derivación al especialista hasta lograr un diagnóstico definitivo4,5.
En cambio, el cribado neonatal es un procedimiento aplicado a los recién nacidos poco después del parto, usualmente durante las primeras 24 a 48 horas de vida. Su objetivo es descubrir enfermedades genéticas o metabólicas en sus primeras etapas, incluso antes de que se manifiesten síntomas. Mediante una sencilla muestra de sangre extraída del talón del bebé, se buscan indicios de ciertas EERR. Si los resultados del cribado son positivos o indican una posible enfermedad, se procede a realizar pruebas diagnósticas adicionales para confirmar el diagnóstico y proporcionar el tratamiento adecuado de manera oportuna6.

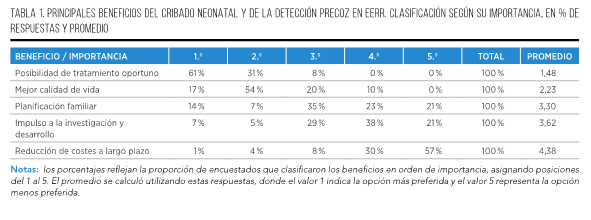
La detección temprana de EERR presenta una serie de beneficios significativos tanto para los pacientes como para el sistema de salud en general. Algunos de estos beneficios incluyen:
- Tratamiento en etapas iniciales: Un diagnóstico temprano permite iniciar el tratamiento adecuado lo antes posible, mejorando significativamente la calidad de vida del paciente y, en ocasiones, siendo vital para evitar el deterioro de la salud y prevenir complicaciones graves7.
- Reducción de costes: La detección precoz de una enfermedad poco frecuente puede ayudar a evitar procedimientos médicos innecesarios y tratamientos inapropiados, lo que a su vez puede disminuir los costes de la atención médica tanto para el paciente como para el sistema de salud en general8,9.
- Impulso a la investigación y desarrollo de tratamientos: Un diagnóstico en fases iniciales permite una mejor comprensión de las EERR, lo que estimula la investigación científica y el desarrollo de nuevos tratamientos para abordar estas condiciones de una manera más efectiva10.
- Apoyo emocional y psicosocial: La identificación temprana de una EERR brinda a los pacientes y sus familias la oportunidad de poder conectarse con grupos de apoyo y comunidades que se enfrentan a desafíos similares, muchas veces a través de asociaciones de pacientes11. También les permite acceder a apoyo psicológico y social de manera más temprana, lo cual resulta fundamental para abordar la afectación emocional, aumentar la autoestima, desarrollar mejores estrategias de afrontamiento y elevar el bienestar psicológico de las familias afectadas12.
- Planificación familiar: Detectar tempranamente una EERR posibilita una mejor planificación familiar y la toma de decisiones fundamentadas sobre reproducción y aspectos hereditarios, incluyendo futuros embarazos, la realización de pruebas genéticas prenatales y recibir asesoramiento especializado, entre otras opciones13.
DETECCIÓN PRECOZ: DESAFÍOS, AVANCES Y PROPUESTAS DE MEJORA
Desafíos en la detección precoz de EERR
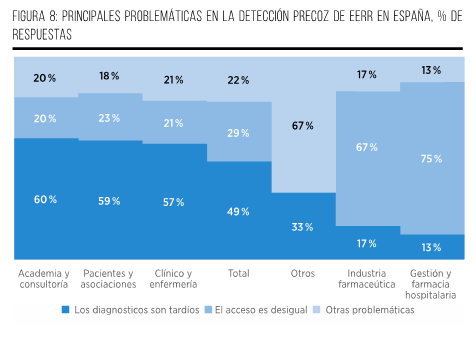
La detección temprana y precisa de las EERR plantea retos importantes. En este contexto, el tiempo medio de diagnóstico de dichas afecciones asciende a 6,18 años en España. Aproximadamente el 57 % de las personas que padecen estas patologías se enfrentan a una espera de más de un año para obtener un diagnóstico definitivo. De este grupo, el 19 % sufre una demora de 1 a 3 años, el 17 % experimenta periodos de 4 a 9 años sin tener claridad sobre su condición y, lo que es especialmente alarmante, un 21 % tienen que esperar más de 10 años para conseguir un diagnóstico adecuado2.
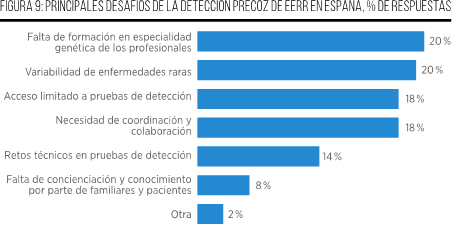
Esta problemática ha sido estudiada por el Grupo de Trabajo de Naciones en Desarrollo (DNWG) del Programa Internacional de Enfermedades No Diagnosticadas (UDNI) en 20 países, incluyendo España. Los resultados revelan que la limitación de financiación para pruebas y servicios genéticos, así como la falta de fondos para investigaciones multidisciplinarias, son algunos de los principales factores que ralentizan el proceso de detección y diagnóstico14.
El cribado neonatal y la detección temprana son cruciales para abordar el impacto de las enfermedades raras
Además, la falta de conciencia sobre las EERR tanto en la población general como en la comunidad médica representa un obstáculo significativo, llevando a demoras en buscar atención médica y a diagnósticos erróneos. Asimismo, la falta de coordinación entre programas de salud e instituciones genera ineficiencias en el diagnóstico de pacientes con EERR14.
Por su parte, otros factores asociados con el retraso diagnóstico han sido identificados en un estudio con pacientes del Registro de Pacientes con Enfermedades Raras de España. Entre ellos, figuran la necesidad de viajar para consultar a especialistas diferentes, visitar a una gran cantidad de profesionales clínicos, ser diagnosticado en una región diferente a la de residencia y padecer una EERR del sistema nerviosoI. En términos del tiempo requerido para consultar a un especialista, un periodo de espera de más de 6 meses para ser derivado desde la primera visita médica fue lo que más contribuyó al retraso en el diagnóstico5,15.

Avances científicos que han mejorado las opciones diagnósticas
Estos datos y desafíos resaltan la urgente necesidad de mejorar los procesos de detección y diagnóstico de las EERR. A pesar de ello, es importante destacar que, en los últimos años, se han logrado avances científicos relevantes en el ámbito de la detección de EERR, lo que ha permitido una mejora sustancial en las opciones diagnósticas disponibles para las personas que sufren estas afecciones.
Una de las principales estrategias que ha revolucionado el diagnóstico genético de las EERR es la secuenciación de exomas y genomas. Esta técnica ha demostrado ser altamente efectiva en la identificación de causas genéticas subyacentes a diversas afecciones, como anomalías congénitas y retrasos en el desarrollo o discapacidad intelectual. Gracias a la secuenciación de exomas, se ha logrado un rendimiento diagnóstico superior al 50 % en algunos casos con presentación clínica bien definida. Además, analizar de forma combinada al paciente y a sus progenitores ha aumentado la precisión del diagnóstico, permitiendo una identificación más certera de las variantes genéticas causantes de la enfermedad17–19.
Además de analizar los genes y su secuencia, también se están utilizando otras técnicas especiales llamadas «ómicas» para comprender mejor las EERR. Estos enfoques incluyen el estudio de cómo los genes se activan y expresan (transcriptómica), cómo ciertas modificaciones químicas afectan a los genes (epigenómica), qué proteínas están presentes en el cuerpo (proteómica) y qué pequeñas moléculas están involucradas en los procesos metabólicos (metabolómica). Estas herramientas proporcionan información adicional que puede ayudar a resolver casos complicados cuando la secuencia del ADN no ha dado respuestas claras17,20,21.
En algunos estudios, el rendimiento diagnóstico se ha visto mejorado entre un 7,5 % y 18 % mediante el análisis complementario de transcriptomas y exomas (otras técnicas “ómicas”)21–24. Además, el desarrollo de nuevos ensayos y técnicas, como el análisis del metiloma para trastornos de la improntaII y las cromatinopatíasIII, ha abierto nuevas posibilidades para el diagnóstico de enfermedades poco comunes17,27.

Soluciones propuestas para diagnósticos tempranos y eficaces
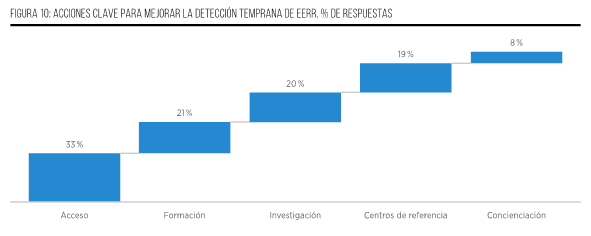
A pesar de los notables avances científicos, es imperativo continuar impulsando el desarrollo de soluciones que permitan realizar diagnósticos tempranos y eficaces de EERR. Las siguientes propuestas buscan mejorar el enfoque en este campo5,14,29,30:
- Desarrollar estrategias específicas de salud pública adaptadas a las particularidades de cada país para abordar los desafíos relacionados con el diagnóstico de EERR.
- Garantizar una financiación adecuada para pruebas genéticas y herramientas de diagnóstico que permitan una detección temprana y precisa de las EERR.
- Fomentar el intercambio de datos, la investigación clínica y la experiencia en diagnósticos entre diferentes países, lo que puede enriquecer la comprensión y el abordaje de estas afecciones.
- Establecer centros especializados y multidisciplinarios de referencia para el diagnóstico genómico, donde los expertos puedan colaborar y compartir conocimientos para mejorar los procesos de diagnóstico temprano.
- Impulsar el uso de inteligencia artificial y enfoques científicos avanzados, como los mencionados en el apartado anterior (y nuevas técnicas que puedan surgir), para complementar las herramientas de diagnóstico y aumentar la precisión en la identificación de EERR.
- Realizar reanálisis periódicos de los datos genómicos para descubrir nuevas asociaciones entre genes y enfermedades, lo que puede llevar a nuevos avances en el diagnóstico.
- Mejorar la eficiencia del sistema de salud, abordando los tiempos de espera y simplificando los procesos de derivación.
- Aumentar la concienciación sobre las EERR entre los profesionales de la salud y los pacientes para mejorar la detección temprana y el manejo adecuado de estas enfermedades.
- Empoderar a los pacientes y sus familias, proporcionando herramientas y recursos que les permitan hacer preguntas relevantes durante la búsqueda de soluciones médicas.
- Incorporar la temática de EERR en los planes de estudio de medicina y en la formación de profesionales de la salud, lo que contribuirá a una mayor sensibilización y capacidad de abordaje de estas patologías.
La detección temprana mejora la calidad de vida, reduce costes e impulsa la investigación en enfermedades raras
CRIBADO NEONATAL: SITUACIÓN ACTUAL, AVANCES, DESAFÍOS Y RECOMENDACIONES POLÍTICAS
Situación actual en España y Europa
El cribado neonatal representa uno de los programas más trascendentales para identificar precozmente afecciones de carácter infrecuente. En España, este procedimiento está regulado por nuestra legislación, siendo considerado tanto una medida de salud pública, orientada a preservar el bienestar de la sociedad en su conjunto, como un servicio de atención médica de vital importancia31.
En nuestro país, el Programa de Cribado Neonatal (PCN) incluido en la cartera común de servicios asistenciales del Sistema Nacional de Salud (SNS) abarca siete enfermedades, las cuales se ofrecen a todos los recién nacidos: hipotiroidismo congénito (HC), fenilcetonuria (PKU), fibrosis quística (FQ), deficiencia de acil-coenzima A-deshidrogenasa de cadena media (MCADD), deficiencia de 3-hidroxi-acil-coenzima A-deshidrogenasa de cadena larga (LCHADD), acidemia glutárica tipo I (GA-I) y anemia falciforme (AF)32,33. Sin embargo, en comparación con otros países europeos como Italia (48), Polonia, Austria o Portugal (todos con 29), entre otros, el número de enfermedades incorporadas en el PCN nacional español es considerablemente inferior, tal y como se muestra en la Figura 232.
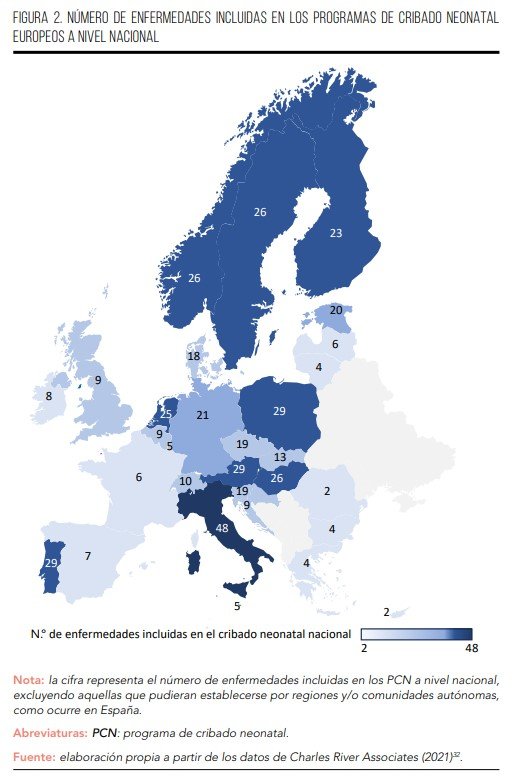 La inclusión de estas siete enfermedades en el PCN fue respaldada por el Consejo Interterritorial del SNS en julio de 201334. Para tomar esta decisión, se tuvieron en cuenta criterios esenciales para cada enfermedad, su implementación en programas ya establecidos en las comunidades autónomas, informes sobre su eficacia clínica, análisis de coste-efectividad (cuando estos estuvieran disponibles), el grado de recomendación basado en evidencia científica y el consenso del grupo de trabajo y de un comité de ética35. Su desarrollo e implementación implican la colaboración de las autoridades sanitarias a nivel nacional y regional31.
La inclusión de estas siete enfermedades en el PCN fue respaldada por el Consejo Interterritorial del SNS en julio de 201334. Para tomar esta decisión, se tuvieron en cuenta criterios esenciales para cada enfermedad, su implementación en programas ya establecidos en las comunidades autónomas, informes sobre su eficacia clínica, análisis de coste-efectividad (cuando estos estuvieran disponibles), el grado de recomendación basado en evidencia científica y el consenso del grupo de trabajo y de un comité de ética35. Su desarrollo e implementación implican la colaboración de las autoridades sanitarias a nivel nacional y regional31.
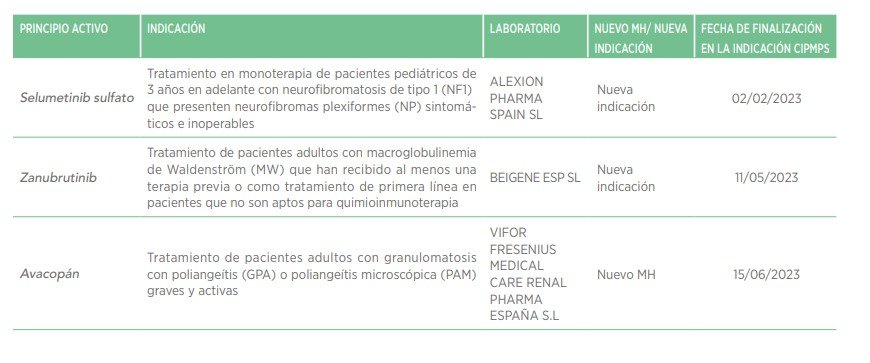
En febrero de 2023, el Ministerio de Sanidad anunció que en el futuro se incluirán cuatro patologías endocrino-metabólicas (déficit de biotinidasa, enfermedad de la orina con olor a jarabe de arce, homocistinuria, e hiperplasia suprarrenal congénita) en el PCN, además de incorporar el cribado de hipoacusia y programas de cribado prenatal para anomalías cromosómicas y enfermedades infecciosas36–38.
En los últimos años, se ha observado una ampliación de los PCN en Europa, incrementándose el número total de enfermedades evaluadas mediante el cribado. Este fenómeno se debe, en parte, a la creciente adopción de tecnologías que permiten el tamizaje neonatal, como el uso de la espectrometría de masas en tándem. Además, el notable aumento de tratamientos disponibles, incluidas terapias génicas, ha ampliado las opciones terapéuticas para EERR que anteriormente carecían de soluciones adecuadas32.
Acceso a los programas de cribado en España
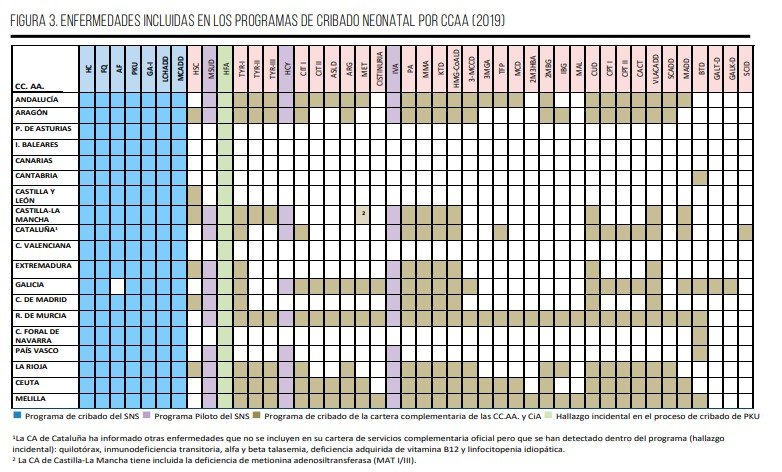
En España, dada la descentralización de las competencias sanitarias, los programas de cribado no solo se incorporan a nivel nacional, sino también desde el ámbito regional. Así, además de las siete enfermedades incluidas en el PCN de la cartera común básica de servicios asistenciales del SNS, trece comunidades autónomas y las dos ciudades autónomas han incorporado oficialmente otras enfermedades a sus PCN, como parte de sus respectivas carteras de servicios complementarias. Así, regiones como Andalucía, Aragón, Cataluña, Galicia, La Rioja, Ceuta o Melilla han implementado pruebas para más de 30 enfermedades, mientras que otras, como Cantabria y Asturias, solo cubren ocho32,39, lo que en la práctica implica diferencias significativas en la cobertura de enfermedades en España.
Superar barreras, como la falta de financiación y la falta de concienciación, es esencial para mejorar el diagnóstico temprano y el cribado neonatal
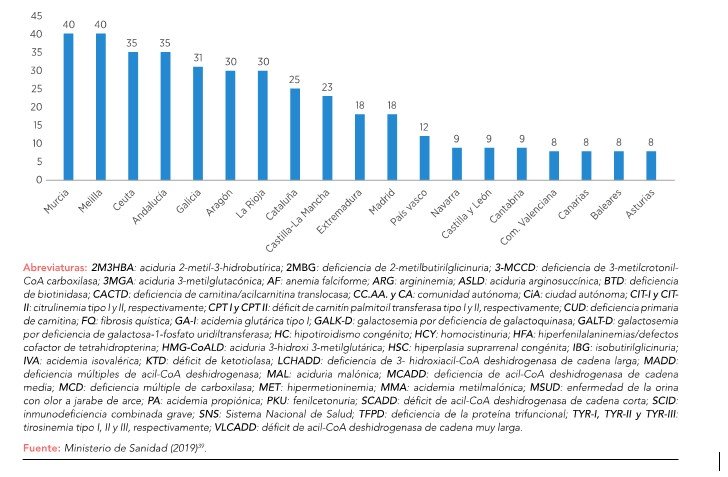
En nuestro país, son objeto de cribado un total de 40 enfermedades metabólicas, tal y como se muestra en la Figura 339. Algunas enfermedades, como la inmunodeficiencia combinada grave (SCID, Severe Combined Inmunodeficiency) y las galactosemias no se criban en prácticamente ninguna de las comunidades autónomas, lo que supone un problema a la hora de establecer su diagnóstico precoz39.
Ante esta situación de inequidad en el acceso a los PCN, la Federación Española de Enfermedades Raras (FEDER) aboga por incorporar en la cartera de servicios del SNS todas aquellas enfermedades que cumplan con los criterios de efectividad clínica y eficiencia, así como con los requerimientos éticos y legales para la implantación de los programas de cribado neonatalVI. Esto se basa en la experiencia y éxito de casos previamente incorporados, respaldados por evidencias generadas por programas autonómicos de cribado y colaboración entre las distintas comunidades41.
Barreras, mejores prácticas y recomendaciones políticas
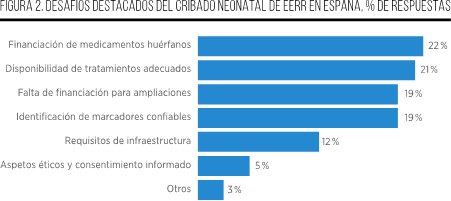
En el ámbito de la expansión e implementación de los PCN, las políticas nacionales desempeñan un papel impulsor fundamental. Sin embargo, en algunos países, a pesar de la declaración de intenciones por parte de los responsables políticos de mejorar y ampliar estos programas, se ha observado una limitación o incluso la falta de avances significativos en los últimos años. Aunque las acciones gubernamentales son de gran importancia, existen diversas barreras que deben ser superadas para facilitar la implementación de los diferentes PCN, incluyendo el mayor número de EERR a diagnosticar de manera precoz.
Entre estas barreras, destacan el complicado proceso de proponer la ampliación de los paneles de enfermedades a incluir, así como los requisitos de infraestructura que limitan el ritmo de expansión de los PCN y la disponibilidad de laboratorios y personal capacitado. Además, se presentan obstáculos como la variación en la implementación del programa una vez aprobado, la falta de financiación para las ampliaciones, y la ausencia de criterios comunes para demostrar el valor del cribado32.
Para superar estas barreras y facilitar la expansión de los PCN, se han identificado buenas prácticas que podrían ser de gran ayuda. En base a esto, se han desarrollado recomendaciones de políticas, clasificadas en función de tres procesos fundamentales identificados32,41:
Factores que afectan a las propuestas de inclusión de enfermedades:
- Se debe aplicar un proceso de revisión periódica de las enfermedades ya incluidas en los PCN para evaluar si su perfil de beneficio-riesgo sigue siendo favorable y si alguna enfermedad ya no debería formar parte del programa.
- La participación de representantes profesionales de pacientes (por ejemplo, grupos de defensa de pacientes y expertos clínicos) puede impulsar el apoyo político para la creación de comités nacionales de cribado y permitir la expansión del PCN.
Factores que afectan a la revisión y aprobación de la inclusión de enfermedades en los PCN:
1. Los organismos responsables de las decisiones deben ser transparentes al exponer sus procesos, criterios de evaluación y requisitos en materia de pruebas para facilitar la presentación de propuestas de ampliación de los paneles.
2. Estos mismos organismos deberían garantizar la armonización de sus criterios para promover procesos de evaluación eficientes.
3. Las autoridades deben consultar a los representantes de pacientes y médicos en cada fase del proceso de ampliación para evaluar la inclusión de cualquier enfermedad adicional en el panel de cribado.
4. Las evaluaciones deben tener en cuenta la disponibilidad y el beneficio clínico de los tratamientos pertinentes a la hora de evaluar el valor potencial de una prueba de cribado para una enfermedad específica.
Factores que afectan a la implementación:
- Se deben realizar ejercicios de mapeo de las ubicaciones y capacidades actuales de los laboratorios, para garantizar que las futuras actualizaciones de los PCN se implementen de manera oportuna.
- Es necesario establecer objetivos en torno a los plazos de implementación de programas aprobados.
En conclusión, las EERR representan un desafío significativo para la salud pública debido a su diversidad y dificultades en el diagnóstico temprano. Los retrasos en la identificación de estas patologías generan sufrimiento a los pacientes y sus familias, además de imponer un alto coste al sistema de salud. Por ello, la detección temprana y el cribado neonatal desempeñan un papel crucial en el abordaje de estas afecciones. La identificación temprana permite un tratamiento oportuno, reduce los costes y estimula el desarrollo de terapias dirigidas y avances en la investigación clínica. La aplicación de estrategias como la secuenciación de exomas y genomas ha mejorado significativamente las opciones diagnósticas, y las técnicas «ómicas» están brindando información valiosa para resolver casos complejos. Para mejorar la detección temprana de las EERR, es necesario implementar políticas que garanticen una financiación adecuada, fomentar el intercambio de datos y la investigación clínica, establecer centros especializados y multidisciplinarios, y empoderar a los pacientes y sus familias con recursos y apoyo. Por su parte, el cribado neonatal constituye una herramienta crucial para detectar enfermedades genéticas o metabólicas en recién nacidos antes de que se manifiesten síntomas. En España y resto de Europa, los PCN han avanzado, pero todavía existen disparidades en la cantidad de enfermedades incluidas en los paneles. La expansión y mejora de estos programas requieren políticas gubernamentales claras, un proceso transparente y abierto para proponer modificaciones, así como la participación de partes interesadas y la adopción de criterios claros para justificar la inclusión de una enfermedad en el cribado. La implementación de estas recomendaciones políticas allanará el camino hacia un diagnóstico temprano y preciso de las EERR, lo que tendrá un impacto positivo en la calidad de vida de los pacientes y sus familias.
I. Las personas afectadas por enfermedades del sistema nervioso muestran un mayor riesgo de experimentar retrasos, debido al hecho de que muchas de estas enfermedades tienen una gran complejidad diagnóstica y que muchas de sus manifestaciones clínicas se superponen, lo que requiere pruebas más específicas para llegar a un diagnóstico específico de la enfermedad.
II. Trastornos de la impronta: Son enfermedades genéticas raras que ocurren debido a cambios en las marcas químicas de ciertos genes. Estas marcas, llamadas «improntas», juegan un papel importante en cómo funcionan los genes. Cuando hay cambios inusuales en estas marcas, puede causar problemas en el desarrollo y funcionamiento del cuerpo25.
III. Cromatinopatías: Son condiciones médicas que ocurren debido a alteraciones en la estructura de la cromatina. La cromatina es la forma en que el ADN se organiza y empaqueta en el núcleo de las células. Cuando hay problemas en la estructura de la cromatina, puede afectar cómo se activan y desactivan los genes, lo que puede llevar a diversas enfermedades y trastornos26.
IV. El ARN (ácido ribonucleico) es una molécula que transfiere y traduce la información genética para la síntesis de proteínas.
V. La metilación es un proceso epigenético que consiste en la adición de grupos metilo (-CH3 ) a ciertas bases de ADN o proteínas, afectando la expresión génica sin alterar la secuencia del ADN.
VI. Los criterios que debe satisfacer una enfermedad para ser incluida en un programa de detección precoz neonatal financiado por el Sistema Público de Salud son: (1) La enfermedad cursa con morbilidad mental o física severa y/o mortalidad si no se diagnostica en el periodo neonatal. (2) La búsqueda clínica mediante un simple examen físico no es efectiva y no identifica la enfermedad en este periodo. (3) Existe un tratamiento efectivo disponible. (4) El tratamiento precoz mejora significativamente el pronóstico. (5) La enfermedad tiene una incidencia relativamente elevada: > 1 por 10.000-15.000 recién nacidos. (6) Existe un test analítico de cribado, rápido, sencillo, fiable y de bajo coste40.
REFERENCIAS
- Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020 Feb;28(2):165–73.
- Benito-Lozano J, López-Villalba B, Arias-Merino G, Posada de la Paz M, Alonso-Ferreira V. Diagnostic delay in rare diseases: data from the Spanish rare diseases patient registry. Orphanet J Rare Dis. 2022;17(1):418.
- Giugliani R, Castillo Taucher S, Hafez S, Oliveira JB, Rico-Restrepo M, Rozenfeld P, et al. Opportunities and challenges for newborn screening and early diagnosis of rare diseases in Latin America. Front Genet. 2022; 13. Disponible en: https://www.frontiersin.org/articles/10.3389/fgene.2022.1053559
- Weller D, Vedsted P, Rubin G, Walter FM, Emery J, Scott S, et al. The Aarhus statement: improving design and reporting of studies on early cancer diagnosis. Br J Cancer. 2012;106(7):1262–7.
- Benito-Lozano J, Arias-Merino G, Gómez-Martínez M, Ancochea-Díaz A, Aparicio-García A, Posada de la Paz M, et al. Diagnostic Process in Rare Diseases: Determinants Associated with Diagnostic Delay. Int J Environ Res Public Health. 2022;19(11):6456.
- Asociación Española de Pediatría. ¿Qué es el cribado neonatal o prueba del talón? Disponible en: https://www.aeped.es/rss/en-familia/que-es-cribado-neonatal-o-prueba-talon
- Bogart K, Hemmesch A, Barnes E, Blissenbach T, Beisang A, Engel P, et al. Healthcare access, satisfaction, and health-related quality of life among children and adults with rare diseases. Orphanet J Rare Dis. 2022;17(1):196.
- Chung CCY, Ng NYT, Ng YNC, Lui ACY, Fung JLF, Chan MCY, et al. Socio-economic costs of rare diseases and the risk of financial hardship: a cross-sectional study. Lancet Reg Health – West Pac. 2023; 34. Disponible en: https://www.thelancet.com/journals/lanwpc/article/PIIS2666-6065(23)00029-9/fulltext
- Garrison S, Kennedy A, Manetto N, Pariser AR, Rutter JL, Yang G. The Economic Burden Of Rare Diseases: Quantifying The Sizeable Collective Burden And Offering Solutions. Health Aff Forefr 2022; Disponible en: https://www.healthaffairs.org/do/10.1377/forefront.20220128.987667/full/
- Tesi B, Boileau C, Boycott KM, Canaud G, Caulfield M, Choukair D, et al. Precision medicine in rare diseases: What is next? J Intern Med. 2023 Oct;294(4):397-412.
- FEDER. Declaración Institucional: Campaña “Haz que el tiempo vaya a nuestro favor”. 2023. Disponible en: https://www.enfermedades-raras.org/actualidad/posicionamientos/declaracion-institucional-campana-haz-que-el-tiempo-vaya-nuestro-favor
- Kenny T, Stone J. Psychological Support at Diagnosis of a Rare Disease: a review of the literature. Rare Disease Research Partners. 2022. Disponible en: https://rd-rp.com/wp-content/uploads/2022/03/220202-Literature-Review-Report.pdf
- Gee M, Piercy H, Machaczek K. Family planning decisions for parents of children with a rare genetic condition: A scoping review. Sex Reprod Healthc Off J Swed Assoc Midwives. 2017;14:1–6.
- Taruscio D, Salvatore M, Lumaka A, Carta C, Cellai LL, Ferrari G, et al. Undiagnosed diseases: Needs and opportunities in 20 countries participating in the Undiagnosed Diseases Network International. Front Public Health. 2023;11:1079601.
- FEDER. El Proyecto ‘DetERminantes del retraso diagnóstico’ identifica las causas y consecuencias de la odisea diagnóstica en enfermedades raras. 2023. Disponible en: https://www.enfermedades-raras.org/actualidad/noticias/el-proyecto-determinantes-del-retraso-diagnostico-identifica-las-causas-y-consecuencias-de-la-odisea-diagnostica-en-enfermedades-raras
- Genome4care.com. El caso de Delfina, la niña que llevaba 10 años sin conocer la causa de su enfermedad 2019. Disponible en: https://www.genome4.com/care/el-caso-de-delfina-la-nina-que-llevaba-10-anos-sin-conocer-la-causa-de-su-enfermedad/
- Tolosa A. Avances en el diagnóstico de las enfermedades raras. Genotipia. Genética Médica News. 2023. Disponible en: https://genotipia.com/genetica_medica_news/avances-en-el-diagnostico-de-las-enfermedades-raras/
- Boycott KM, Hartley T, Kernohan KD, Dyment DA, Howley H, Innes AM, et al. Care4Rare Canada: Outcomes from a decade of network science for rare disease gene discovery. Am J Hum Genet. 2022;109(11):1947–59.
- Casas-Alba D, Hoenicka J, Vilanova-Adell A, Vega-Hanna L, Pijuan J, Palau F. Diagnostic strategies in patients with undiagnosed and rare diseases. J Transl Genet Genomics. 2022;6(3):null.
- Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med Off J Am Coll Med Genet. 2021;23(11):2029–37.
- Colin E, Duffourd Y, Tisserant E, Relator R, Bruel AL, Tran Mau-Them F, et al. OMIXCARE: OMICS technologies solved about 33% of the patients with heterogeneous rare neuro-developmental disorders and negative exome sequencing results and identified 13% additional candidate variants. Front Cell Dev Biol. 2022;10:1021785.
- Murdock DR, Dai H, Burrage LC, Rosenfeld JA, Ketkar S, Müller MF, et al. Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J Clin Invest. 2021;131(1):e 141500.
- Lee H, Huang AY, Wang L kai, Yoon AJ, Renteria G, Eskin A, et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet Med Off J Am Coll Med Genet. 2020;22(3):490–9.
- Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9(386):eaal5209.
- Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tümer Z, Monk D, et al. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics. 2015;7:123.
- Di Fede E, Grazioli P, Lettieri A, Parodi C, Castiglioni S, Taci E, et al. Epigenetic disorders: Lessons from the animals–animal models in chromatinopathies. Front Cell Dev Biol. 2022; 10. Disponible en: https://www.frontiersin.org/articles/10.3389/fcell.2022.979512
- Sadikovic B, Levy MA, Kerkhof J, Aref-Eshghi E, Schenkel L, Stuart A, et al. Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med Off J Am Coll Med Genet. 2021;23(6):1065–74.
- Pinazo-Durán MD. Genética y algo más. Arch Soc Espanola Oftalmol. 2012;87(2):35–7.
- Marwaha S, Knowles JW, Ashley EA. A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Med. 2022;14(1):23.
- Global Commission. Ending the Diagnostic Odyssey for Children with a Rare Disease. Takeda, Microsoft, EURORDIS; 2018 p. 36. Disponible en: https://globalrarediseasecommission.com/Report/assets/static/documents/GlobalCommission-print-021919-a68c8ce2a5.pdf
- Nicolás Jiménez MP, Pàmpols Ros T, García López FJ, Martín Arribas C, Pérez Aytés A, García Sagredo JM, et al. Medio siglo de cribado neonatal en España: evolución de los aspectos éticos, legales y sociales (AELS). Parte II, marco legal. 2021; Disponible en: http://addi.ehu.es/handle/10810/55170
- Charles River Associates. A landscape assessment of newborn screening (NBS) in Europe. 2021. Disponible en: https://media.crai.com/wp-content/uploads/2021/11/19130322/CRA-LS-Insights-NBS-Policy.pdf
- Ministerio de Sanidad. Programas de cribado neonatal. 2023. Disponible en: https://www.sanidad.gob.es/profesionales/saludPublica/prevPromocion/Cribado/cribadoNeonatal.htm
- Redacción Médica. El Ministerio pacta una cartera única de cribados neonatales. 2013. Disponible en: https://www.redaccionmedica.com/noticia/el-ministerio-pacta-una-cartera-unica-de-cribados-neonatales-1435
- Ministerio de Sanidad, Servicios Sociales e igualdad. Resumen ejecutivo del grupo de expertos sobre concreción de cartera común de servicios para cribado neonatal. 2013. Disponible en: https://www.sanidad.gob.es/profesionales/saludPublica/prevPromocion/Cribado/docs/ResumenEjecutivoCribadoNeonatal.pdf
- Diariofarma. Sanidad saca a consulta la actualización de la cartera de servicios. 18 de febrero. 2022. Disponible en: https://diariofarma.com/2022/02/18/sanidad-saca-a-consulta-la-actualizacion-de-la-cartera-de-servicios
- Ministerio de Sanidad. Proyecto de Orden por la que se modifican los anexos I, II, III, VI y VII del Real Decreto 1030/2006, de 15 de septiembre, por el que se establece la cartera de servicios comunes del Sistema Nacional de Salud y el procedimiento para su actualización. 17 de febrero. 2022. Disponible en: https://www.sanidad.gob.es/normativa/audiencia/docs/PRD_cartera_de_servicios_comunes_SNS.pdf
- Infosalus. Sanidad incluirá cuatro enfermedades raras más en el cribado neonatal. Europa Press; 2023. Disponible en: https://www.infosalus.com/actualidad/noticia-sanidad-incluira-cuatro-enfermedades-raras-mas-cribado-neonatal-20230228135017.html
- Ministerio de Sanidad. Programa de cribado neonatal del Sistema Nacional de Salud. Informe de evaluación. 2019. Disponible en: https://www.sanidad.gob.es/profesionales/saludPublica/prevPromocion/Cribado/docs/InformeDeEvaluacionSICN_2019.pdf
- Calderón-López GM, Jiménez-Parrilla F, Losada-Martínez A. Screening neonatal. Asociación Española de Pediatría. 2008. Disponible en: https://www.aeped.es/sites/default/files/documentos/44.pdf
- FEDER. Homogeneizar y ampliar los programas de cribado neonatal, objetivo prioritario de FEDER y el tejido asociativo de enfermedades raras este 2023. 2023. Disponible en: https://www.enfermedades-raras.org/actualidad/noticias/homogeneizar-y-ampliar-los-programas-de-cribado-neonatal-objetivo-prioritario-de-feder-y-el-tejido-asociativo-de-enfermedades-raras-este-2023

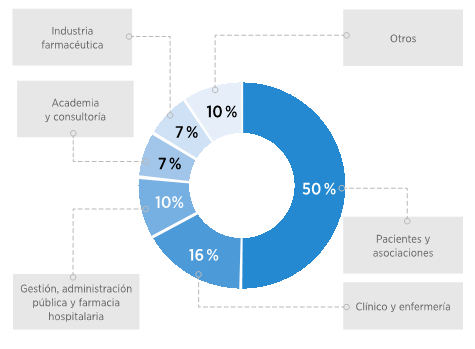
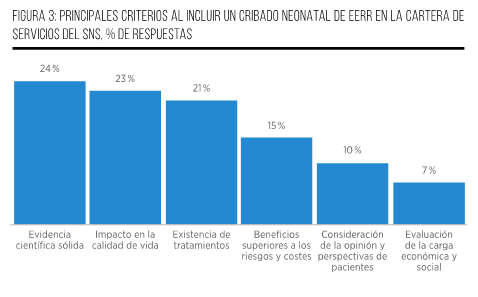
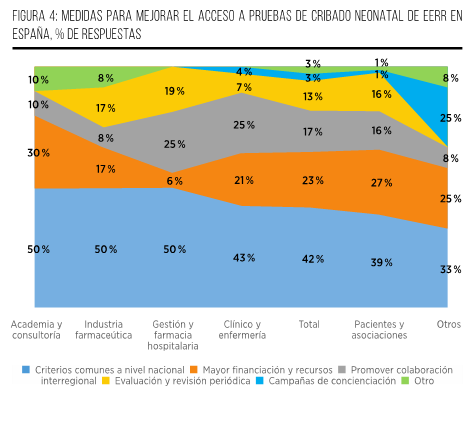
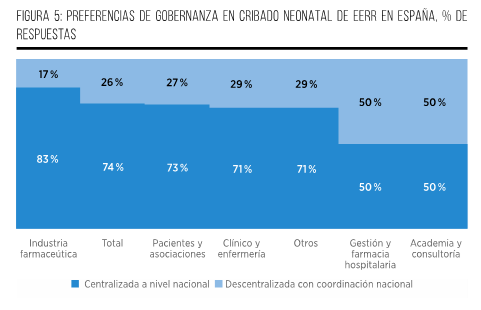
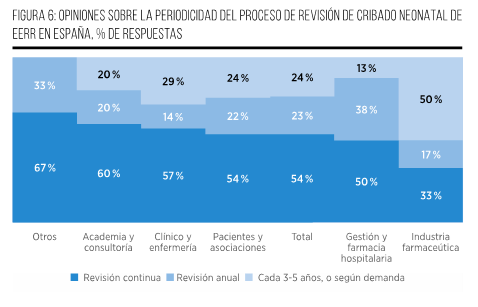
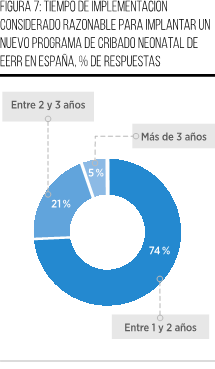 Finalmente, el 74 % de los participantes considera que, una vez que un PCN sea aprobado, su implementación debería llevarse a cabo en un plazo de entre 1 y 2 años. Esta opinión se fundamenta en consideraciones relacionadas con la adecuada planificación, establecimiento de protocolos y realización de pruebas piloto, considerando este período como un tiempo razonable y eficiente. Por otro lado, una parte del grupo (21 %) sostiene que la implementación debería llevarse a cabo en un plazo más prolongado, de entre 2 y 3 años, con el objetivo de permitir una planificación más detallada y la definición precisa de los elementos fundamentales del programa. Esto incluiría el diseño y los componentes necesarios para su ejecución, con miras a garantizar su éxito y efectividad. Por último, únicamente un 5 % de los participantes aboga por un plazo de más de 3 años para la implementación del PCN. Sus argumentos se centran en la necesidad de abordar la complejidad de la infraestructura requerida y la integración de tecnologías avanzadas. Consideran que un enfoque gradual sería más adecuado para asegurar la eficiencia y la efectividad del proceso (Figura 7).
Finalmente, el 74 % de los participantes considera que, una vez que un PCN sea aprobado, su implementación debería llevarse a cabo en un plazo de entre 1 y 2 años. Esta opinión se fundamenta en consideraciones relacionadas con la adecuada planificación, establecimiento de protocolos y realización de pruebas piloto, considerando este período como un tiempo razonable y eficiente. Por otro lado, una parte del grupo (21 %) sostiene que la implementación debería llevarse a cabo en un plazo más prolongado, de entre 2 y 3 años, con el objetivo de permitir una planificación más detallada y la definición precisa de los elementos fundamentales del programa. Esto incluiría el diseño y los componentes necesarios para su ejecución, con miras a garantizar su éxito y efectividad. Por último, únicamente un 5 % de los participantes aboga por un plazo de más de 3 años para la implementación del PCN. Sus argumentos se centran en la necesidad de abordar la complejidad de la infraestructura requerida y la integración de tecnologías avanzadas. Consideran que un enfoque gradual sería más adecuado para asegurar la eficiencia y la efectividad del proceso (Figura 7).