
Los medicamentos de terapia avanzada (advanced therapy medicinal products, ATMP) y los dirigidos al tratamiento de las enfermedades raras (EERR) generalmente producen mejoras muy significativas en la salud y calidad de vida de los pacientes, suponiendo incluso, en algunos casos, la cura de enfermedades que antes carecían de alternativas terapéuticas, en estadios muy avanzados.
El objetivo de las administraciones públicas sanitarias en España es asegurar a los ciudadanos “el derecho a la protección de la salud, con el objetivo común de garantizar la equidad, la calidad y la participación social en el SNS” 1. Parte del cumplimiento de este objetivo pasa por la gestión del acceso al mercado de los medicamentos. Por ello, conviene entender los elementos esenciales considerados en este proceso, particularmente para las terapias dirigidas a las EERR y los ATMP.
A lo largo del artículo revisaremos el contexto en el que se presentan estos fármacos y los principales criterios y mecanismos que forman parte de la actual estructura de acceso. Además, analizaremos el impacto de algunos instrumentos técnico-normativos implementados recientemente, y presentaremos elementos que, añadidos a dicha estructura, podrían potencialmente beneficiar el proceso para acceder a estos medicamentos.
Definiciones y particularidades de las terapias avanzadas en el ámbito de las enfermedades raras
Las EERR se caracterizan por ser patologías de baja prevalencia y con características particulares. Muchas de ellas debutan en la infancia, son crónicas y degenerativas, potencialmente mortales y conllevan un alto impacto en la calidad de vida del paciente y sus cuidadores2,3. Algunos países las definen según su prevalencia relativa, que a su vez también varía dependiendo de la población del país. En Europa, una enfermedad se considera rara cuando afecta a menos de 1 de cada 2.000 habitantes2.
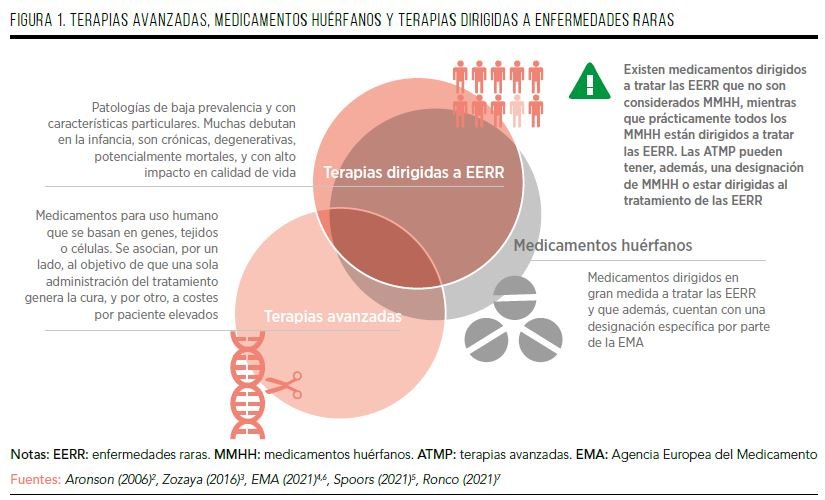
Por su parte, los ATMP son medicamentos para uso humano que se basan en genes (terapia génica), tejidos (terapia tisular) o células (terapia celular). Su objetivo es curar la enfermedad con una única administración del tratamiento (“once and done”), si bien su contrapartida son unos costes por paciente elevados4,5. Las terapias dirigidas a EERR son todas aquellas que, teniendo o no una designación oficial de “huérfana” por parte de las agencias regulatorias, están destinadas al tratamiento de este tipo de enfermedades poco frecuentes. Los medicamentos huérfanos (MMHH) son aquellos dirigidos a tratar las EERR, y que además cuentan con esta designación específica de “huérfano” (orphan) por parte de la Agencia Europea del Medicamento (EMA), por tratarse de medicamentos sin alternativas terapéuticas. A menudo, los MMHH se asocian a una baja probabilidad de generación de rendimientos, lo que justifica la inversión necesaria para su desarrollo y la generación de beneficios clínicos significativos en el tratamiento, la prevención o diagnóstico de una enfermedad con necesidades no cubiertas de tratamiento, que ponga en peligro la vida o sea crónicamente debilitante6.
Cabe así tener en cuenta algunos posibles solapamientos entre las ATMP, las terapias dirigidas al tratamiento de las EERR y los MMHH. Por un lado, existen medicamentos dirigidos a tratar las EERR que no son considerados MMHH, mientras que prácticamente todos los MMHH están dirigidos a tratar las EERR. Por otro lado, las ATMP pueden tener, además, una designación de MMHH7, o estar dirigidas al tratamiento de una EERR, mientras que otras ATMP pueden no destinarse al tratamiento de las EERR (FIGURA 1).
Evolución e impacto de políticas y mecanismos de fomento
El acceso al mercado de los ATMP y de los fármacos dirigidos a las EERR ha sido incentivado en Europa y España a través de distintas políticas y mecanismos de fomento. El principal marco normativo europeo relacionado con las EERR fue el reglamento Nº 141/2000 de MMHH, cuyo objetivo era establecer un procedimiento comunitario para declarar determinados medicamentos como MMHH, y establecer incentivos para fomentar la investigación, el desarrollo y la comercialización de los medicamentos declarados como tales8.
Por su parte, el marco normativo más importante en Europa en ATMP es el reglamento Nº 1394/2007 que estableció normas específicas para la autorización, supervisión y farmacovigilancia de los ATMP9. En 2009, la EMA creó un comité especializado, el Comité de Terapias Avanzadas (CAT), responsable de la elaboración de un documento con su recomendación al Comité de Medicamentos de Uso Humano (CHMP) de la EMA respecto a cualquier ATMP, para que este pueda, a partir de entonces, preparar su recomendación final4.
Así mismo, a lo largo de las últimas dos décadas, el desarrollo de los medicamentos dirigidos a EERR, los MMHH y los ATMP también se ha beneficiado indirectamente de la introducción, por parte de la EMA, de algunos instrumentos ideados para facilitar el acceso a medicamentos considerados como de gran interés para la salud pública o que, por razones técnicas o éticas, no pudieran aportar toda la evidencia requerida en otras vías de aprobación. Dichos instrumentos incluyen:
la evaluación acelerada, que reduce el tiempo de evaluación de un fármaco en 2 meses (de 210 a 150 días); la autorización condicional, basada en datos menos completos de eficacia y seguridad, renovándose anualmente; la autorización bajo circunstancias excepcionales, basada en la imposibilidad de aportar evidencia completa, ya sea por cuestiones de factibilidad o éticas; y el PRIME, que se basa en un diálogo temprano con los promotores de medicamentos innovadores para optimizar los planes de desarrollo y la evaluación de los mismos, acelerando su acceso10–12.
El acceso al mercado de los ATMP y de los fármacos dirigidos a las EERR ha sido incentivado en Europa y España a través de distintas políticas y mecanismos de fomento.
El esfuerzo realizado en términos normativos sirvió de referencia a los distintos Estados miembros para llevar a cabo medidas específicas de impulso a estos tipos de medicamentos. En nuestro país, pese a no existir una legislación específica sobre terapias dirigidas a EERR, se han legislado aspectos que les afectan particularmente. Entre ellos, cabe destacar el artículo 92 del RD-legislativo 1/201513, que incluye las necesidades específicas de ciertos colectivos como uno de los criterios de financiación del Sistema Nacional de Salud (SNS).
En España, la legislación de ATMP es una trasposición de las directivas europeas que cubre todos los aspectos regulatorios, salvo los de los ATMP de fabricación no industrial, que son específicos de cada país miembro. El RD 477/2014 regula esta autorización, estableciendo los requisitos y garantías que deben cumplir los ATMP de fabricación no industrial para obtener la correspondiente autorización por parte la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), así como los requisitos de trazabilidad y farmacovigilancia una vez han sido autorizados14. Para los procesos regulatorios nacionales se creó el Comité Español de Terapias Avanzadas de la AEMPS15.
A todo ello se añaden los planes estratégicos dedicados específicamente a dar directrices en los ámbitos de las EERR y de las ATMP. El marco estratégico de las EERR se ha definido en España a través de la Estrategia en Enfermedades Raras del SNS, publicada en 2009, y actualizada en 201416. Además de la estrategia nacional, muchas Comunidades Autónomas (CCAA) han incluido medidas específicas para EERR en sus planes de salud17. Por su parte, el Consejo Interterritorial del SNS aprobó el 15 de noviembre de 2018 el “Plan de abordaje de las terapias avanzadas en el Sistema Nacional de Salud: medicamentos receptores de antígenos quiméricos (CAR)”. El objetivo del plan era organizar de forma planificada, equitativa, segura y eficiente la utilización de los medicamentos CAR en el SNS e impulsar la investigación pública y la fabricación propia y pública de estos medicamentos en el ámbito académico del SNS, en unas condiciones que garanticen los estándares de calidad, seguridad y eficacia18.
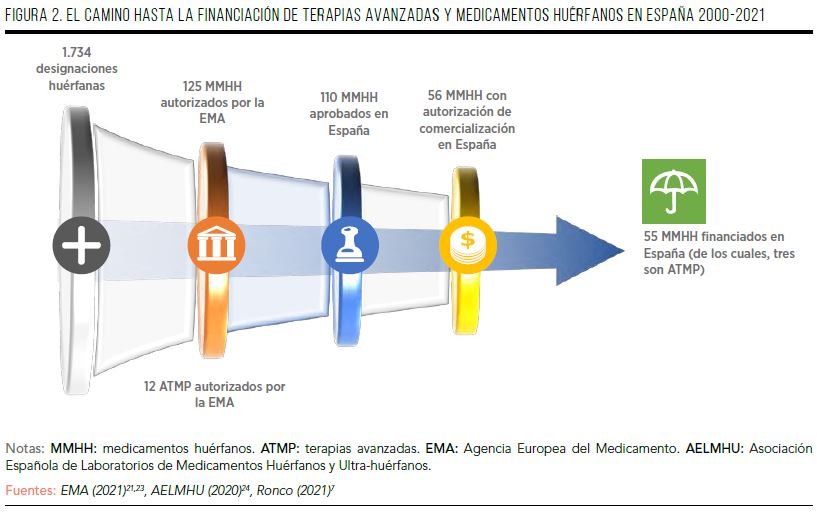
Estas regulaciones, incentivos y políticas de fomento han tenido un claro impacto en diversas áreas relacionadas con los fármacos dirigidos a EERR y las ATMP. En cuanto a las EERR, se ha producido un sustancial aumento en el número de fármacos dirigidos a tratarlas. Entre 2000 y septiembre de 2021, la EMA aprobó 1.734 designaciones huérfanas y había aprobado la comercialización de 125 MMHH, frente a los solo 8 MMHH aprobados antes del año 200019–22. Por su parte, desde 2009 hasta febrero de 2021, la EMA había autorizado 17 ATMP, de las cuales 10 eran terapias génicas, 4 terapias tisulares y 3 terapias celulares23. De estos 17 ATMP, 5 han sido revoca- dos posteriormente, quedando un total de 12 ATMP autorizados actualmente (9 terapias génicas, 2 tisulares y 1 celular)23.
Pese a esta notable evolución en términos de aprobaciones a nivel europeo, España ocupa posiciones intermedias en la UE tanto en grado de acceso como en tiempo de acceso a las terapias. De los 125 MMHH que actualmente disponen de aprobación por parte de la EMA, 56 (un 45%) cuentan con autorización de comercialización en nuestro país (frente a 60-80% en Italia y Francia, y más del 90% en Alemania). A su vez, 55 (un 44%) se encuentran financiados por el SNS24. En cuanto a tiempos de acceso, la mediana de tiempo trascurrido entre la aprobación de la EMA y la disponibilidad del fármaco en España es de 486 días, muy superior a países como Dinamarca, Austria, Finlandia e Italia (109-308 días) o Alemania (reembolso automático)25.
En cuanto a los ATMP, de las 12 terapias actualmente con autorización por parte de la EMA, 3 (25%) han recibido financiación en España (todas ellas con designación de MMHH: 2 terapias génicas CAR-T y 1 terapia celular), 4 (33%) no han recibido financiación (2 terapias tisulares y 2 génicas) y las 5 restantes (41%) están todavía en proceso de decisión de financiación7. En otros países, el número de terapias con financiación es de 9 (75%) en Alemania, 7 (58%) en Reino Unido y 5 (41%) en Italia y Francia (FIGURA 2)7.
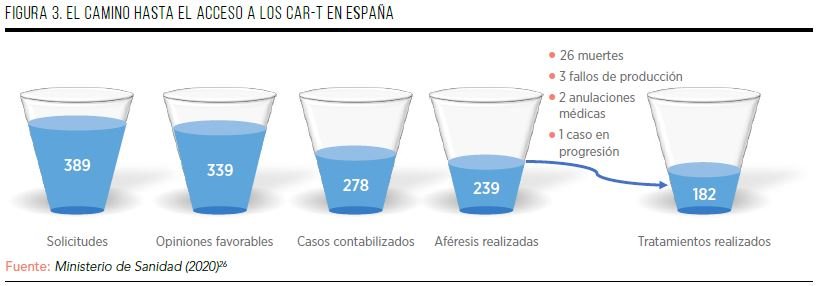
Respecto a la implementación de los CAR-T en nuestro país, entre marzo de 2019 y octubre de 2020 (plan se seguimiento del Ministerio de Sanidad) se recibieron 386 solicitudes de tratamiento, representando una media de 19,3 solicitudes mensuales26. Cada caso fue valorado por expertos del SNS, que dieron un parecer desfavorable a 43 casos (11%) y desestimaron otros 4 (1%). De las 339 solicitudes favorables (88%), se dispone de información de 278 casos, sobre los que se conoce la fecha de realización de la aféresis en 239 pacientes26. De estos 239 pacientes, en 35 casos (15%) no se produjo la administración del CAR-T, siendo el principal motivo la muerte del paciente (26 casos; 74%), seguido de 3 fallos de producción, 2 anulaciones por decisión médica y 1 caso que todavía estaba en progresión. En conclusión, se han comunicado la realización de 182 tratamientos con CAR-T (el 76% de los casos con aféresis conocida, y el 47% de las solicitudes realizadas), con una media de 9 tratamientos mensuales en todo el territorio nacional. En el informe de seguimiento del Ministerio se señala que el periodo de estado de alarma causado por la Covid-19 no afectó a la tendencia en el número de solicitudes, aféresis y tratamientos (FIGURA 3)26.
Elementos esenciales de acceso: la experiencia española y prácticas internacionales
El proceso de entrada de todo medicamento en el mercado europeo se compone de distintas etapas, que pasan por la investigación básica, preclínica y clínica, la evaluación clínica, la autorización de comercialización, la evaluación económica, la decisión de precio y reembolso, la comercialización y el seguimiento post-autorización27,28. Por lo tanto, parte del proceso de evaluación y acceso de las ATMP y de las terapias dirigidas a EERR se realiza a nivel europeo, mientras que otra parte corre a cargo de cada Estado miembro (FIGURA 4).

A continuación, repasamos los elementos vigentes en España y otros países de nuestro entorno.
Generalmente, para la financiación de los ATMP y de las terapias dirigidas a EERR, los países tienen en cuenta distintos criterios, que se pueden agrupar en tres grandes bloques. Por un lado, los criterios clínicos, que son los más utilizados en la práctica habitual. Por otro lado, los criterios económicos relativos al impacto presupuestario, coste de oportunidad, o sostenibilidad del sistema sanitario en su conjunto. Finalmente, a menudo se consideran también en la decisión los denominados criterios humanísticos, que se basan en los conceptos de equidad y justicia, contraponiendo la visión igualitaria a la de maximización de recursos adoptada por cada país, al incluir elementos como la “regla de rescate” como un imperativo moral a la hora de salvar una vida que está en peligro29.
ESPAÑA
En España, el proceso de financiación y acceso a nivel nacional, válido para cualquier medicamento, empieza con la solicitud de Código Nacional por parte del promotor. Una vez que el solicitante comunica la intención de comercializar la terapia en España, la AEMPS elabora un Informe de Posicionamiento Terapéutico (IPT), que servirá como apoyo para definir la resolución de precio y financiación30.
Según el artículo 92 del RD-legislativo 1/2015, de 24 de julio13, la inclusión de cualquier medicamento en la financiación del SNS se posibilita teniendo en cuenta seis criterios, que son la gravedad de la enfermedad, las necesidades no cubiertas, el valor social y terapéutico de la terapia, la racionalización del gasto público, el grado de innovación y la existencia de terapias alternativas31,32. Los dos criterios más utilizados por el Comité Interministerial de Precios de Medicamentos y Productos Sanitarios (CIPM) para la emisión de recomendaciones positivas a medicamentos para EERR y ATMP son la gravedad de la enfermedad y el beneficio social y terapéutico de la terapia33.
En España, la financiación de cualquier medicamento se posibilita teniendo en cuenta seis criterios, de los cuales los más utilizados son la gravedad de la enfermedad y el beneficio social y terapéutico de la terapia.
Los ATMP y los medicamentos dirigidos a las EERR suelen estar asociados a unos elevados costes por paciente, aunque el colectivo al que van dirigidos sea pequeño35. En este sentido, se hace patente la necesidad de implementar nuevos enfoques que ofrezcan a los pagadores un esquema que mitigue las complicaciones asociadas a la financiación de estas terapias, a la vez que facilite el rápido acceso a los pacientes que las necesiten, garantizando al mismo tiempos la sostenibilidad financiera del sistema35. Entre los acuerdos que puede realizar el laboratorio con los reguladores sanitarios figuran el modelo de fijación de techo de gasto para periodos de tiempo determinados; el coste máximo por paciente; los precios variables en función del volumen de compra y los acuerdos de riesgo compartido (ARC) o de pago por resultados en salud36.
La proliferación de estos esquemas de financiación ha ido a la par del crecimiento de las terapias dirigidas a EERR. Desde el año 2000, los pagadores nacionales han aumentado su interés acerca de que los precios pagados se ajusten a los beneficios logrados como medio para ayudar a asignar recursos limitados de forma más eficiente37. Pese a no haber información concreta para España, un estudio realizado en siete países europeos (Holanda, Inglaterra, Gales, Suecia, Alemania, Francia e Italia) observó que la mayoría de los esquemas de financiación utilizados para MMHH se basaron en resultados (55%), frente a esquemas de acuerdos financieros (45%)38.
Por su parte, los CAR-T, que pertenecen a la categoría de terapias génicas de los ATMP, se han financiado en España en base a esquemas de pago por resultados, según los cuales entre el 36% y 56% del pago total por paciente se realizaba al inicio del tratamiento y el restante 18 meses después en función de los datos de supervivencia o de respuesta completa alcanzados7,39. Su introducción se asoció a la implementación de mecanismos de seguimiento que permitieran observar la eficacia y seguridad a largo plazo y en base a la práctica clínica real.
La aprobación de los ATMP y de muchos de los fármacos dirigidos a las EERR está condicionada a la creación de registros40. De hecho, el Plan de Abordaje de las Terapias Avanzadas del Ministerio de Sanidad obliga el uso de VALTERMED para el registro de datos de la práctica clínica real de estos medicamentos18. Sin embargo, el desarrollo de registros para la recopilación de datos en vida real a menudo conlleva ciertas limitaciones, como el tipo de requisitos impuestos, la falta de incorporación de pacientes y otros agentes en su diseño, la falta de información sobre su utilidad real o la existencia de conflictos de intereses en su generación y abordaje41. Se han creado registros específicos para los ATMP y las terapias destinadas a EERR. Algunos de los ejemplos más destacables son el Registro Estatal de EERR y el registro de EERR del Instituto de Salud Carlos III42. Además, desde 2019 está operativo VALTERMED, el sistema de información corporativo del SNS, cuyo objetivo es disponer de información para la adecuada toma de decisiones relacionadas con la prestación farmacéutica, determinando el valor terapéutico en la práctica clínica real de los medicamentos que se utilizan por el sistema público43. Actualmente, existen 11 protocolos farmacoclínicos introducidos en este sistema, siete de los cuales son para terapias dirigidas a las EERR (3 de ellos son ATMP)44.
PRÁCTICAS INTERNACIONALES
Los ATMP, los fármacos dirigidos a las EERR y los MMHH se aprueban de manera centralizada para toda la UE, pero son los Estados miembro los que deciden su comercialización y financiación pública en el país, encontrándose por tanto diferencias en el acceso a estas terapias. En este sentido, cabe mencionar algunas prácticas que realizan países de nuestro entorno para facilitar la financiación y acceso a estas terapias, como por ejemplo la consideración de otros criterios para su financiación, el establecimiento de modelos alternativos de evaluación y financiación, o la utilización de fondos específicos (FIGURA 5).
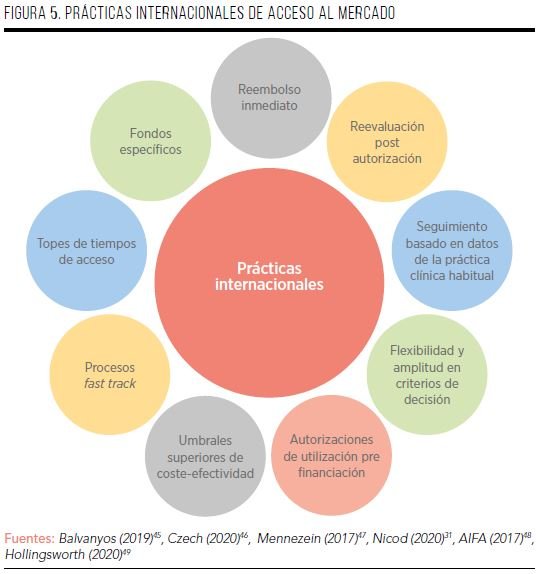
En el caso de las terapias dirigidas a las EERR, cabe destacar algunos hitos en Alemania, Francia, Inglaterra e Italia. Alemania es el único país de Europa que permite el reembolso inmediato de cualquier medicamento, en base a las recomendaciones del Comité de Medicamentos Huérfanos (COMP), lo que beneficia indirectamente a las terapias dirigidas a EERR (y también a los ATMP). A los 6 meses, se reevalúan estos fármacos en cuanto a su eficacia en la práctica clínica habitual. A este proceso le sigue una renegociación del precio que puede durar unos 6 meses más. Si el volumen de ventas del fármaco supera los 50 millones de euros anuales, se aplica el nuevo precio. Además, los MMHH no necesitan de un comparador para la determinación de su valor añadido, y este país adopta también una mayor flexibilidad en relación a los criterios de decisión, valorando la rareza de la enfermedad, o si el fármaco va destinado a una población específica (por ejemplo, niños)31,45,46,50,51.
En Francia, la evaluación de los medicamentos se realiza a través de la asignación del beneficio terapéutico (SMR, que consta de 4 niveles y 5 criterios) y valor terapéutico añadido del fármaco (ASMR, que es un sistema de 5 niveles, donde el nivel I representa el mayor valor añadido) frente a los tratamientos disponibles. Estos criterios tienden a beneficiar a las terapias dirigidas a EERR. De hecho, entre 2000 y 2006, el 40% de los MMHH recibieron un ASMR de entre I y III, en comparación con el 3% del total de fármacos aprobados en el país47. Por su parte, el 70% de los MMHH utilizan un mecanismo denominado autorización temporal de utilización (ATU), que permite el acceso de un fármaco antes de la decisión de precio y financiación46.
En Inglaterra existen algunos mecanismos que, añadidos al proceso habitual de evaluación de cualquier fármaco, benefician directa e indirectamente a los medicamentos dirigidos a las EERR. Uno de ellos es la implementación de un proceso fast-track a los casos en los que el impacto presupuestario no supere los 20 millones de libras anuales22. Por su parte, para los medicamentos ultra-huérfanos (prevalencia de <1/50.000 habitantes), se establecen plazos máximos de acceso (desde la aprobación de la EMA) de 20 meses para los nuevos medicamentos y de 15 meses para las nuevas indicaciones52. Además, si estas terapias demuestran beneficios significativos en términos de años de vida ajustados por calidad (AVAC), el NICE permite umbrales de hasta 300.000 libras por AVAC ganado para su aprobación, lo que es 10 veces superior al umbral estándar aplicado en el país53. Finalmente, las terapias oncológicas disponen de otra vía de acceso, el Fondo de Medicamentos contra el Cáncer, con criterios más amplios de valor, como la aplicación de la perspectiva social31.
Revisión de los instrumentos técnico-normativos más recientes en el contexto actual
Pese a la notable evolución en términos de acceso al mercado de los ATMP y las terapias dirigidas a EERR de los últimos años, queda camino por recorrer. Conscientes de ello, recientemente las autoridades tanto nacionales como europeas han dado algunos pasos, revisando o proponiendo revisar algunos instrumentos técnico-normativos.
En el ámbito nacional, el Ministerio de Sanidad presentó en noviembre de 2020 un plan de consolidación de los IPT para mejorar su proceso de evaluación mediante cambios en la gobernanza y en las herramientas aplicadas en su elaboración56. El objetivo de este instrumento, introducido en 2013, es reducir las desigualdades de acceso entre regiones, al proporcionar un mayor grado de coordinación y cooperación57. El plan se ha desarrollado en torno a las siguientes líneas de actuación (FIGURA 6)58.

- La creación de la red de evaluación de medicamentos (REval- MED), formada por más de 120 expertos en torno a un grupo de evaluación terapéutica, un grupo de evaluación económica y siete nodos de evaluación por áreas clínicas, uno de los cuales es para EERR no oncológicas y terapias La gobernanza de REvalMED se realizará a través de la Comisión Permanente de Farmacia, formada por la Dirección General de Cartera Básica de Servicios del SNS y Farmacia (DGCCSSNSF), por la AEMPS y por representantes de las CCAA56.
- La modificación de la metodología empleada para el diseño y aprobación de los IPT. Se adopta una matriz de priorización que pueda agilizar el proceso de evaluación del medicamentos, basada en cinco criterios58.
1
Lugar en la terapéutica: se asignan 10 puntos si el medicamento cubre una laguna terapéutica en una patología grave; 5 puntos si cubre una laguna en una patología no grave; y 0 puntos si no cubre ninguna laguna terapéutica.
2
Potencial beneficio clínico incremental respecto a alternativas terapéuticas: si existe, se asignan 10 puntos al fármaco; si solo existe en algún subgrupo, se asignan 5 puntos; si no hay beneficio incremental, 0 puntos.
3
Beneficio clínico y seguridad: 5 puntos si el fármaco tuviera un beneficio clínico similar al de las alternativas, pero con un perfil de seguridad mucho mejor; 0 puntos si tanto el beneficio clínico como el perfil de seguridad son similares.
4
Nuevas indicaciones: 10 puntos si se trata de una nueva indicación en medicamentos ya financiados y comercializados; 0 puntos en otro caso.
5
Potencial interés del fármaco para el SNS respecto a las alter- nativas terapéuticas: en este caso, se utiliza una escala de 0 a 20, donde cero representa una relevancia nula y 20, la mayor relevancia posible.
- Tiempos. Se establecen 12 pasos específicos para que los informes se realicen en un plazo estipulado de aproximadamente 90-100 días. Este proceso sigue un procedimiento de 3 fases, en las que las sociedades científicas, las asociaciones de pacientes y la industria pueden realizar comentarios y alegaciones58.
- Desarrollo de un cuadro de mando para el control y seguimiento de los IPT, con indicadores relativos al tiempo de cada etapa y la categorización de los medicamentos posicionados59.
- Inclusión obligatoria de la evaluación económica en los informes, siguiendo la metodología establecida por el Grupo GENESIS60. La perspectiva utilizada será la del financiador sanitario, si bien existe la opción de incluir información sobre la perspectiva social cuando las características de la patología generen diferencias relevantes respecto a la perspectiva sanitaria60. En el caso de las evaluaciones de coste-utilidad, se deben reportar los resultados en euros por AVAC Sin embargo, no se establecen umbrales que puedan considerarse razonablemente aceptables. Además de la evaluación económica, se debe presentar un análisis del impacto presupuestario que conllevaría la implementación del nuevo fármaco en su población diana60.
Dado que el plan de consolidación está todavía en una fase de arranque, todavía no es posible evaluar sus resultados. En todo caso, ya hay un debate acerca del impacto que podría tener el plan sobre el acceso a los ATMP y a las terapias dirigidas a EERR. Algunos opinan que mejorará la coordinación y el consenso sobre los criterios utilizados para el acceso, introduciendo una mayor rapidez y rigor científico al mismo y minimizando las desigualdades en el acceso. Por el contrario, otras voces defienden sus dudas sobre cómo se realizarán las evaluaciones y si estas surtirán el efecto deseado en el acceso, alertando de que involucrar a un mayor número de agentes en el proceso puede perjudicar los avances hacia un proceso más ágil61. Otros alegan que el tiempo previsto para la elaboración de la evaluación farmacoeconómica, de 10 días, parece ser insuficiente62.
Recientemente las autoridades nacionales y europeas han revisado o propuesto revisar algunos instrumentos técnico -normativos, en el sentido de mejorar el acceso a las ATMP y a los MMHH
Por su parte, los principales avances en relación a los ATMP desde el lanzamiento del Plan de Terapias Avanzadas en España (con enfoque en los CAR-T), en noviembre de 2018, han sido la aprobación de criterios y estándares para la designación de centros para la utilización de los medicamentos CAR-T; el procedimiento para la valoración de solicitudes de utilización de CAR-T por el grupo de expertos; el procedimiento para la derivación de pacientes a los centros designados; los procedimientos técnicos para la obtención de la muestra para la fabricación de CAR-T; el protocolo clínico para el manejo de efectos adversos graves en pacientes con CAR-T y la convocatoria de concesión de subvenciones para Proyectos de investigación Clínica del Instituto Carlos III, que prioriza las terapias avanzadas y la introducción del primer CAR-T de fabricación no industrial26. Con sus informes de seguimiento, el Ministerio busca rendir cuentas de manera dinámica sobre el acceso real a estas terapias versus el planeado.
En el ámbito europeo, cabe resaltar algunos instrumentos normativos que se encuentran en fase de revisión, con el propósito de asegurar un mejor acceso a las terapias (ya sean ATMP, dirigidas a EERR o medicamentos en general) para los pacientes que las necesiten. Uno de ellos es la adopción, en noviembre de 2020, de la Estrategia Farmacéutica Europea, que propone cuatro objetivos, entre ellos un mayor acceso, a medicamentos asequibles. Además, la Comisión Europea ha iniciado un proceso de reflexión sobre como adaptar el sistema de incentivos para estimular la innovación en áreas con importantes necesidades no cubiertas (que incluyen, por ejemplo, enfermedades neurodegenerativas, raras y cánceres infantiles). Finalmente, se propone establecer una infraestructura común e interoperable, para mejorar el intercambio y acceso a datos sanitarios en la UE63.
Por su parte, se está llevando a cabo una revisión del reglamento europeo Nº 141/2000 sobre MMHH, en el sentido de alinear los incentivos al contexto actual64. Las líneas de actuación abordarán temas como el desequilibrio entre el número de designaciones y aprobaciones de MMHH; la compensación excesiva generada a casi un cuarto de las empresas beneficiarias del periodo de exclusividad de 10 años; y la adecuación del criterios de prevalencia, entre otras65.
Finalmente, en noviembre de 2020 el proyecto RARE2030 propuso ocho recomendaciones para EERR, para servir de referencia a una nueva generación de políticas nacionales y europeas en los próximos 10 años. Estas recomendaciones incluyen una mejor coordinación a nivel europeo, mayor precisión y reducción en los tiempos de diagnóstico, un mayor número de terapias (más fármacos), con mayor acceso (más fármacos financiados) y asequibilidad (menores precios), y una atención equitativa que incluya la atención transfronteriza y virtual, la reducción de la vulnerabilidad psicológica, social y económica, una I+D guiada por las necesidades no cubiertas, y una mejor integración y disponibilidad de datos66.
Propuestas para promover un acceso más ágil y equitativo
REFERENCIAS
- BOE. Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud. 2003.
- Aronson Rare diseases and orphan drugs. Br J Clin Pharmacol. 2006;61(3):243-5, doi: 10.1111/j.1365-2125.2006.02617.x.
- Zozaya N, Villoro, R, Hidalgo, Enfermedades raras: aspectos clave en el entorno actual. newsRARE. 2016;1(1):6-12.
- European Medicines Agency (EMA). Advanced therapy medicinal products: European Medicines Agency. Disponible en: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview.
- Spoors J, Miners A, Cairns J, Palnoch D, Summerfield A, McEntee J, et al. Payer and Implementation Challenges with Advanced Therapy Medicinal Products (ATMPs). BioDrugs Clin Immunother Biopharm Gene 2021;35(1):1-5, doi: 10.1007/s40259-020-00457-4.
- European Medicines Orphan designation: Overview. Disponible en: https://www.ema.europa.eu/en/human-regula- tory/overview/orphan-designation-overview
- Ronco V, Dilecce M, Lanati E, Canonico PL, Jommi C. Price and reimbursement of advanced therapeutic medicinal products in Europe: are assessment and appraisal diverging from expert recommendations? J Pharm Policy Pract. 2021;14(1):30, doi: 10.1186/s40545-021- 00311-0.
- Consejo Europeo, Parlamento Reglamento (CE) no 141/2000. 22 de enero de 2000.
- Parlamento Europeo. Reglamento (CE) No1394/2007 del Parlamento Europeo y del Consejo del 13 de noviembre de 2007 sobre medi- camentos de terapia 2007.
- EMA. Marketing authorisation – Accelerated assessment. Disponible en: http://www.ema.europa.eu/ema/index.jsp?curl=pa-ges/regulation/general/general_content_000955.jsp&mid=WC0b01ac05809f843a.
- EMA. Marketing authorisation – Conditional marketing authorisation. Disponible en: http://www.ema.europa.eu/ema/index.js- p?curl=pages/regulation/general/general_content_000925.jsp&mid=WC0b01ac05809f843b.
- EMA. Exceptional circunstances. Disponible en: https://www.ema.europa.eu/en/glossary/exceptional-circumstances.
- MSSSI. Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios. BOE Num 177. 2015.
- ISCIII – Red de Terapia Celular (TerCel). Libro Blanco de la Terapia Celular en España. Instituto de Salud Carlos III; Ministerio de Economía y Competitividad; Fondo Europep de Desarrollo Regional; 2018.
- AEMPS. Comité Español de Terapias Avanzadas de la AEMPS. 2015, Disponible en: https://www.aemps.gob.es/investigacion- clinica_terapiasavanzadas_comite_ta/.
- Ministerio de Estrategia en Enfermedades Raras del Sistema Nacional de Salud. 2014.
- Federación Española de Enfermedades Raras (FEDER). Mapa de Políticas Sociosanitarias de Enfermedades 2019.
- Ministerio de Sanidad, Consumo y Bienestar Plan de Abordaje de las terapias avanzadas en el sistema nacional de salud: Medicamentos CAR. Aprobado en el Consejo Interterritorial del SNS del 15 de noviembre. 2018.
- Comisión Evaluación conjunta del Reglamento (CE) no 1901/2006 del Parlamento Europeo y del Consejo, de 12 de diciembre de 2006, sobre medicamentos para uso pediátrico, y del Reglamento (CE) no 141/2000 del Parlamento Europeo y del Consejo, de 16 de diciembre de 1999, sobre medicamentos huérfanos. 2020.
- EMA. Orphan medicines figures 2000-2019. 2020.
- EMA. Download medicine data. Disponible en: https://www.ema.europa.eu/en/medicines/download-medicine-data.
- Farmaindustria. El 20% de los ensayos clínicos en España están ya focalizados en enfermedades raras. FarmaIndustria. Disponible en: https://www.farmaindustria.es/web/otra-noticia/el-20-de-los-ensayos-clinicos-en-espana-estan-ya-focalizados-en-en-fermedades-raras/.
- EMA. Timely patient access to Advanced Therapy Medicinal Products (ATMPs). 2021.
- AELMHU. Informe de acceso de los medicamentos huérfanos en España 2020. Disponible en: https://aelmhu.es/index.php/faq/mmhh/item/download/255_0356039374fd3f4cf9c89d73aa149c49.
- IQVIA. EFPIA Patients W.A.I.T. Indicator 2019 Survey. Disponible en: https://www.efpia.eu/media/554526/patients-wait-indicator-2019.pdf.
- Ministerio de Sanidad. Informe de Seguimiento de la Dirección General de Cartera Común de Servicios del Sistema Nacional de Salud (SNS) y Farmacia sobre el Plan para el Abordaje de las Terapias Avanzadas en el 2020.
- EMA. From laboratory to patient: the journey of a medicine assessed by EMA. 2020.
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Cómo se regulan los medicamentos y productos sanitarios en España. 2014.
- Zozaya, N, Villoro, R, Hidalgo, A, Sarria, A. Criterios de financiación y reembolso de los medicamentos huérfanos. Monografía del Insti- tuto de Salud Carlos 2016.
- de Burgos Informes de posicionamiento terapéutico: contexto y retos. El Farm. 2019;375:31-6.
- Nicod E, Whittal A, Drummond M, Facey Are supplemental appraisal/reimbursement processes needed for rare disease treatments? An international comparison of country approaches. Orphanet J Rare Dis. 2020;15(1):189, doi: 10.1186/s13023-020-01462-0.
- Nicod E, Whittal A, Drummond M, Facey Country vignettes on HTA appraisal/reimbursement processes for rare disease treatments. 2020.
- Ministerio de Comisión Interministerial de Precios de Medicamentos y Productos Sanitrios. Disponible en: https://www.mscbs.gob.es/profesionales/farmacia/CIPMyPS.htm.
- Luis-Yagüe Innovación y acceso de medicamentos. Farmaindustria. 2014.
- Zozaya N, Villaseca J, Hidalgo-Vega Á. Proyecto RET-A: Think-Tank sobre Terapias Reflexión estratégica para el manejo e implementación de las nuevas Terapias Avanzadas en España. Fund Weber. 2020.
- AIRef. Anexo 7. Farmacia Hospitalaria. Estudio gasto hospitalario del sistema nacional de salud: farmacia e inversión en bienes de equipo. 2020.
- Piatkiewicz T, Traulsen J, Holm-Larsen Risk-Sharing Agreements in the EU: A Systematic Review of Major Trends. PharmacoEconomics- Open. 2018, doi: 10.1007/s41669-017-0044-1.
- Morel T, Arickx F, Befrits G, Siviero P, van der Meijden C, Xoxi E, et al. Reconciling uncertainty of costs and outcomes with the need for access to orphan medicinal products: a comparative study of managed entry agreements across seven European Orphanet J Rare Dis. 2013;8:198.
- Jørgensen J, Hanna E, Kefalas P. Outcomes-based reimbursement for gene therapies in practice: the experience of recently launched CAR-T cell therapies in major European J Mark Access Health Policy. 2020;8(1):1715536, doi: 10.1080/20016689.2020.1715536.
- Kodra Y, Weinbach J, Posada-de-la-Paz M, Coi A, Lemonnier SL, van Enckevort D, et al. Recommendations for Improving the Quality of Rare Disease Int J Environ Res Public Health. 2018;15(8), doi: 10.3390/ijerph15081644.
- Hollak CEM, Sirrs S, van den Berg S, van der Wel V, Langeveld M, Dekker H, et al. Registries for orphan drugs: generating evidence or marketing tools? Orphanet J Rare 2020;15, doi: 10.1186/s13023-020-01519-0.
- BOE. Real Decreto 1091/2015 de 4 de diciembre, por el que se crea y regula el Registro Estatal de Enfermedades Raras.
- Ministerio de Sanidad, Consumo y Bienestar Preguntas y respuestas frecuentes sobre el sistema de información hará determinar el valor terapéutico en la práctica clínica real de los medicamentos de alto impacto sanitario y económico en el sistema nacional de salud (VALTERMED). 2019.
- Ministerio de Sanidad, Consumo y Bienestar Social. Sistema de Información para determinar el Valor Terapéutico en la Práctica Clínica Real de los Medicamentos de Alto Impacto Sanitario y Económico en el SNS (VALTERMED). Disponible en: https://www.mscbs.es/profesionales/farmacia/valtermed/home.htm.
- Balvanyos J, Nicod E, Hutchings ORPH-VAL Principles in practice: Comparison of alignment of five European P&R systems. 2019.
- Czech M, Baran-Kooiker A, Atikeler K, Demirtshyan M, Gaitova K, Holownia-Voloskova M, et al. A Review of Rare Disease Policies and Orphan Drug Reimbursement Systems in 12 Eurasian Front Public Health. 2019;7:416, doi: 10.3389/fpubh.2019.00416.
- Mennezein L, Avot D, Laigle V. Orphan Drugs In France: Key Market Access Incentives. Value Health. 2017;20(9):A565, doi: 10.1016/j. 2017.08.943.
- AIFA. Criterios para la clasificación de fármacos innovadores y fármacos oncológicos innovadores. Disponible en: http://www.agen-ziafarmaco.gov.it/content/aifa-criteri-la-classificazione-dei-farmaci-innovativi-e-dei-farmaci-oncologici-innovativi.
- Hollingsworth Increasing Patient Access to Advanced Therapies; the UK Perspective. 2020.
- Zozaya González N, Villoro R, Hidalgo Á. Evaluación económica y financiación de los medicamentos huérfanos. 2017;2(3):7- 16.
- Value Dossier. Orphan Drugs | Benefit assessment of Orphan Drugs | Germany. Disponible en: https://www.value-dossier.com/orphan-drugs-drugs-for-rare-diseases.
- Powell, T, O’Donnell NICE appraisals of rare diseases. Debate Pack Number CDP-2019-0022. House of Commons Library. Disponible en: https://researchbriefings.files.parliament.uk/documents/CDP-2019-0022/CDP-2019-0022.pdf.
- Berdud M, Drummond M, Towse Establishing a reasonable price for an orphan drug. Cost Eff Resour Alloc. 2020;18(1):31, doi: 10.1186/ s12962-020-00223-x.
- Fortinguerra F, Tafuri G, Trotta F, Addis Using GRADE methodology to assess innovation of new medicinal products in Italy. Br J Clin Pharmacol. 2020;86(1):93-105, doi: https://doi.org/10.1111/bcp.14138.
- Garattini L, Curto Performance-Based Agreements in Italy: «Trendy Outcomes» or Mere Illusions? PharmacoEconomics Auckl. 2016;34(10):967-9, doi: http://dx.doi.org/10.1007/s40273-016-0420-1.
- Ministerio de Presentación del plan para la consolidación de los Informes de Posicionamiento Terapéutico (IPT) de los medica- mentos en el Sistema Nacional de Salud (SNS). 2020.
- Pulido S. Acceso a medicamentos huérfanos: España mantiene su cuello de botella. EDS – Econ. 2021, Disponible en: https://econo- com/topics/difusion/acceso-a-medicamentos-huerfanos-espana-mantiene-su-cuello-de-botella/.
- Riesgo M. Así serán los nuevos IPTs: evaluación económica y doce procesos para agilizar su aprobación. El Glob. 2020, Disponible en: https://elglobal.es/politica/asi-seran-los-nuevos-ipts-evaluacion-economica-y-doce-procesos-para-agilizar-su-apro- bacion/.
- Ministerio de Plan para la consolidación de los Informes de Posicionamiento Terapéutico de los medicamentos en el Sistema Nacional de Salud. 2020.
- Ortega Eslava A, Marín Gil R, Fraga Fuentes MD, López-Briz E, Puigventós Latorre Guía de evaluación económica e impacto presu- puestario en los informes de evaluación de medicamentos. SEFH. Sociedad Española de Farmacia Hospitalaria; 2016.
- González El futuro de la evaluación de medicamentos en España. Rev Esp Econ Salud. 2021;16(1).
- Abellán JM, Espín J, Mestre J, Oliva Regulación de precios y financiación de nuevos medicamentos: elementos para el debate en España. 2021.
- European Pharmaceutical Strategy for Europe. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions. COM (2020) 761 Final. 2020.
- Markus C, Michaux G, Rasmussen EUROPE – Are Revisions of the Orphan Drug Regulation and Adoption of a Regulation on “Unmet Medical Needs” on the Horizon. JD Supra. 2020.
- European Joint evaluation of Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use and Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 Decem- ber 1999 on orphan medicinal products. Disponible en: https://op.europa.eu/en/publication-detail/-/publication/e9a9fff0- dbd9-11ea-adf7-01aa75ed71a1/language-en.
- Talarico R, Marinello D, Cannizzo S, Gaglioti A, Ticciati S, Carta C, et Shaping the Future of Rare Diseases after a Global Health Emer- gency: Organisational Points to Consider. Int J Environ Res Public Health. 2020;17(22), doi: 10.3390/ijerph17228694.
- ORPHAR-SEFH. Horizon Scanning – Medicamentos Huérfanos. Segundo diciembre de 2020.
- Zlateva Risk sharing agreements: country experiences, challenges and lessons learned. 2018.







